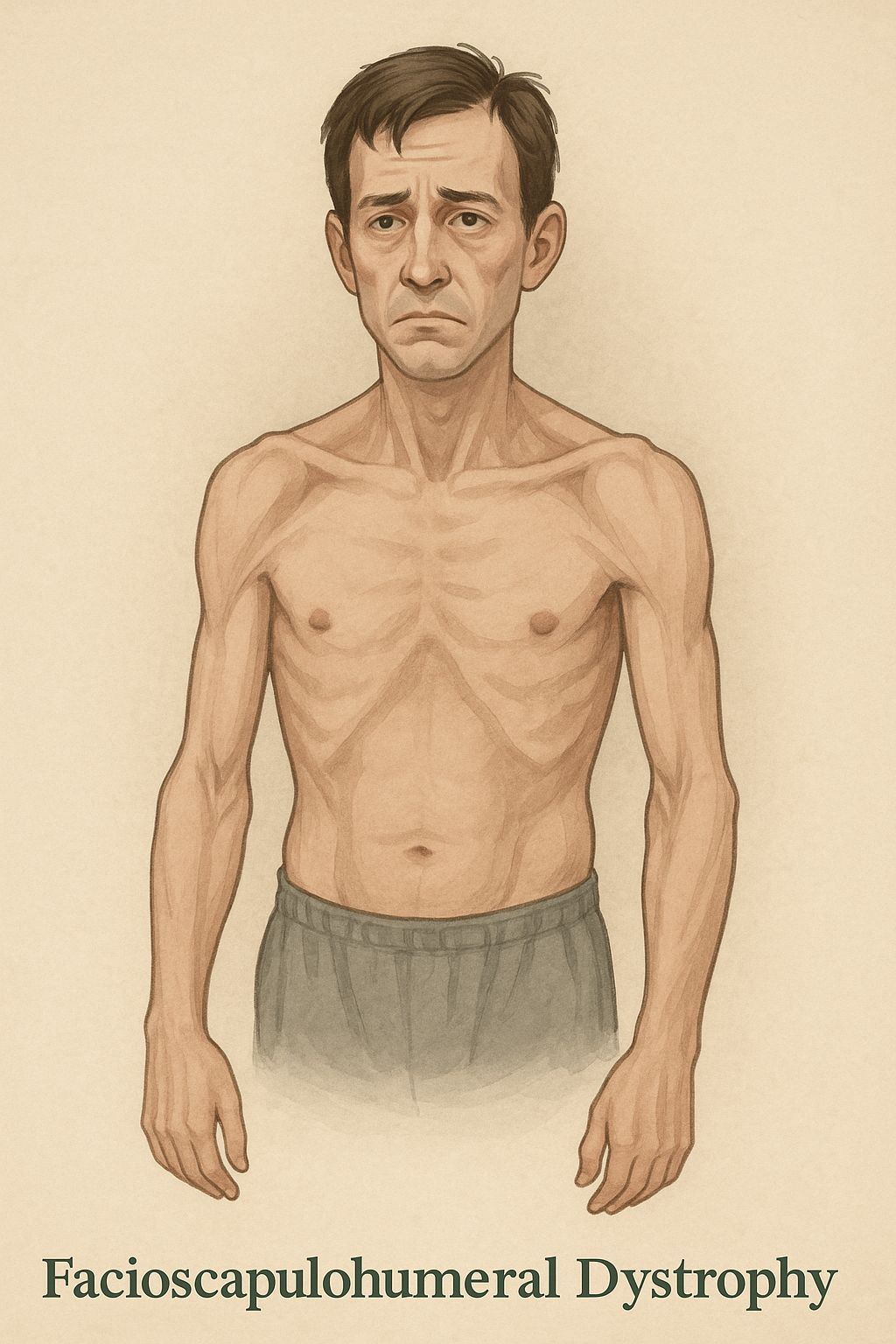
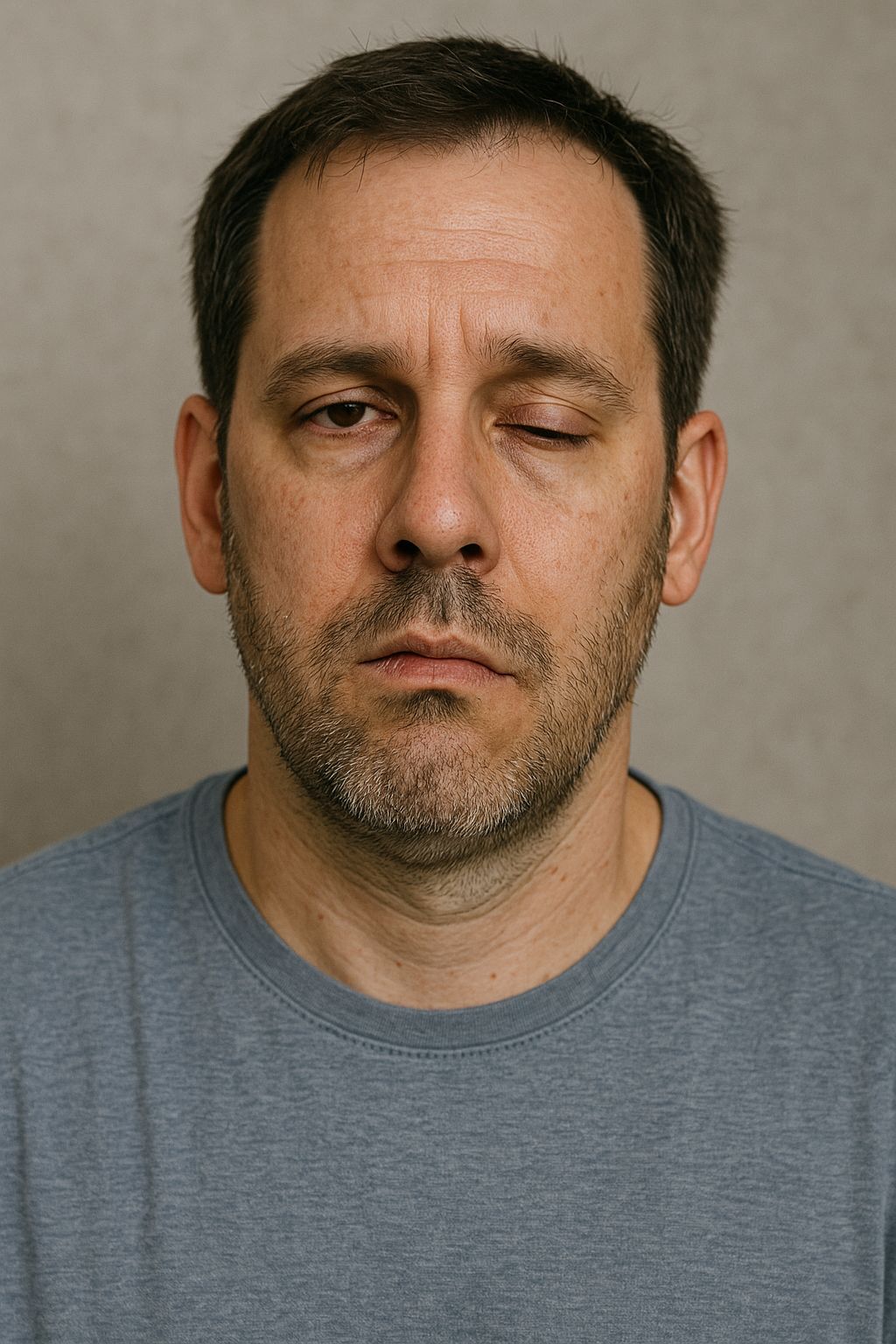
Acute Inflammatory Demyelinating Polyradiculoneuropathy (AIDP)
📊 Quick Facts
- Subtype: AIDP = 80-90% of GBS in North America/Europe
- Incidence: 1-2 per 100,000/year
- Triggers: C. jejuni (40%), CMV, EBV, M. pneumoniae
- Peak Weakness: Median 2 weeks from onset
- Respiratory Failure: 25-30% require mechanical ventilation
⚠️ Red Flags
- Rapidly progressive weakness → Monitor FVC q4-6h
- FVC <20 mL/kg or NIF >-30 cmH₂O → Intubation likely needed
- Autonomic instability → Arrhythmias, BP swings (ICU monitoring)
- Bifacial weakness + ataxia → Miller Fisher syndrome (anti-GQ1b)
- Asymmetric weakness → Consider other diagnosis (not typical GBS)
Clinical Scenario
A 42-year-old man presents with rapidly progressive weakness in both legs over 5 days, now involving the arms and associated with areflexia.
He reports a preceding episode of diarrhea two weeks earlier but denies sensory loss or bowel/bladder symptoms.
Examination reveals symmetrical flaccid weakness, absent deep tendon reflexes, and mild distal paresthesias.
Cranial nerve involvement is evident with bilateral facial weakness, but ocular motility is preserved.
Respiratory function is declining, prompting admission to the intensive care unit for monitoring.
Epidemiology
AIDP is the most common subtype of Guillain-Barré syndrome (GBS) in North America and Europe, representing 80–90% of cases.
The annual incidence of GBS is approximately 1–2 per 100,000, with a slight male predominance and a bimodal age distribution.
The condition often follows infections, particularly with Campylobacter jejuni, cytomegalovirus, Epstein–Barr virus, or Mycoplasma pneumoniae.
Seasonal variation is minimal, but outbreaks may occur following specific infectious exposures.
Although typically sporadic, rare familial clustering suggests a possible genetic predisposition.
Etiopathophysiology
AIDP is an immune-mediated demyelinating polyradiculoneuropathy triggered by molecular mimicry between microbial antigens and peripheral nerve components.
Autoimmune activation leads to macrophage infiltration, complement activation, and segmental demyelination at the level of spinal roots and peripheral nerves.
Conduction block and slowed nerve conduction velocity result from myelin loss, impairing saltatory conduction.
Secondary axonal degeneration may occur in severe or prolonged cases, contributing to residual deficits.
The primary target antigens often involve gangliosides and myelin proteins, though specific autoantibodies are not always detected.
Key Mechanisms: - Molecular mimicry: Microbial antigens (e.g., C. jejuni GM1-like epitopes) resemble peripheral nerve gangliosides - T-cell and B-cell activation: Cross-reactive antibodies target myelin components - Complement-mediated demyelination: MAC (membrane attack complex) formation at nodes of Ranvier - Macrophage infiltration: Stripping and phagocytosis of myelin sheaths - Conduction block: Disruption of saltatory conduction along demyelinated segments
Classic triad of AIDP/GBS: (1) Areflexia (earliest and most consistent finding), (2) Albuminocytologic dissociation on CSF (elevated protein >0.55 g/L with normal WBC <10 cells/μL after week 1), and (3) Ascending symmetric weakness reaching peak by 4 weeks. Always monitor FVC and NIF closely—respiratory failure can develop rapidly even without dyspnea. “The 20/30/40 rule”: FVC <20 mL/kg, NIF >-30 cmH₂O, or SpO₂ <92% suggests impending respiratory failure.
Clinical Features
AIDP typically presents with rapidly progressive, symmetric, ascending weakness beginning in the lower limbs and spreading proximally.
Areflexia is a hallmark finding, while mild distal sensory symptoms such as paresthesias are common but less prominent.
Cranial nerve involvement occurs in up to 50% of patients, most commonly facial nerve palsy, while bulbar weakness can impair swallowing.
Autonomic dysfunction, including cardiac arrhythmias, orthostatic hypotension, and urinary retention, can complicate the course.
Respiratory muscle involvement occurs in approximately 25–30% of patients, necessitating close monitoring of vital capacity.
Diagnosis
The diagnosis is clinical, supported by electrodiagnostic and cerebrospinal fluid (CSF) findings.
Nerve conduction studies reveal prolonged distal latencies, slowed conduction velocity, conduction block, and prolonged F-wave latency.
CSF analysis shows albuminocytologic dissociation (elevated protein with normal cell count) after the first week.
MRI of the spine may show gadolinium enhancement of nerve roots, particularly in the cauda equina.
Differential Diagnosis Comparison:
| Onset |
Acute (hours-4 weeks) |
Acute (hours-4 weeks) |
Acute (hours-days) |
Subacute (weeks-months) |
| Weakness Pattern |
Ascending, symmetric |
Ascending, symmetric |
Legs ± arms (level-dependent) |
Ocular > bulbar > limb |
| Sensory Loss |
Mild distal |
Minimal/absent |
Sensory level present |
None |
| Reflexes |
Areflexia (early) |
Areflexia |
Hyperreflexia (after spinal shock) |
Normal or fatigable |
| Autonomic |
Common (BP/HR swings) |
Common |
Bowel/bladder early |
Rare |
| CSF Protein |
Elevated (>0.55 g/L) |
Elevated |
Variable, may ↑ WBC |
Normal |
| EMG/NCS |
Demyelinating (↓CV, ↑DL, CB) |
Axonal (↓CMAP, normal CV) |
Not done (MRI diagnostic) |
Decremental RNS |
| MRI Spine |
Nerve root enhancement |
Nerve root enhancement |
Cord T2 hyperintensity + enhancement |
Normal |
| Prognosis |
70-80% full recovery |
Variable, may be severe |
Variable |
Treatable, chronic |
AIDP/GBS Diagnosis requires ALL:
Bilateral flaccid weakness of limbs (symmetric)
Decreased or absent deep tendon reflexes in weak limbs
Monophasic illness pattern (peak weakness by 4 weeks)
CSF findings: Protein >0.45 g/L (normal <0.45) with WBC <50 cells/μL
Electrodiagnostic findings: Demyelinating features (prolonged distal latencies, slowed CV, conduction block, prolonged/absent F-waves) in ≥2 nerves
Exclusion criteria: No other identifiable cause (polio, diphtheria, toxins, vasculitis, acute porphyria, critical illness neuropathy)
Management
Treatment Approach (Evidence-Based Recommendations):
1. Immunotherapy (Acute Phase - Within 2 Weeks of Onset):
| IVIG |
0.4 g/kg/day |
5 consecutive days |
Grade A |
Equally effective as PLEX; easier administration; preferred in autonomic instability |
| Plasma Exchange (PLEX) |
200-250 mL/kg total |
4-6 sessions over 10-14 days |
Grade A |
Equally effective as IVIG; requires central line; avoid in cardiovascular instability |
| Corticosteroids |
— |
— |
Grade D (Not recommended) |
Ineffective in GBS; may worsen outcomes |
IVIG vs PLEX Comparison:
| Efficacy |
Equal to PLEX |
Equal to IVIG |
| Administration |
Peripheral IV |
Central line required |
| Duration |
5 days |
10-14 days (4-6 sessions) |
| Cost |
High |
Very high |
| Contraindications |
IgA deficiency, renal impairment |
Cardiovascular instability, coagulopathy |
| Side Effects |
Headache, thrombosis, renal impairment |
Hypotension, bleeding, line infection |
| Preferred When |
Autonomic instability, easier access |
Severe disease, no IVIG contraindications |
2. Respiratory Monitoring & Support:
| FVC |
>70 mL/kg |
<20 mL/kg |
Consider intubation |
| NIF (Negative Inspiratory Force) |
<-60 cmH₂O |
>-30 cmH₂O |
Consider intubation |
| SpO₂ |
>95% |
<92% on room air |
Supplemental O₂ → intubation if worsening |
| Bulbar weakness |
Absent |
Difficulty swallowing/handling secretions |
Aspiration precautions → intubation if severe |
3. Supportive Care & Complication Prevention:
| DVT Prophylaxis |
Enoxaparin 40 mg SC daily or heparin 5,000 U SC TID |
Grade A |
Watch for bleeding if PLEX |
| Pain Management |
Gabapentin 300-1,200 mg TID or carbamazepine 200-400 mg BID |
Grade B |
Neuropathic pain common (85%) |
| Autonomic Monitoring |
Continuous telemetry, BP monitoring |
Grade A |
Arrhythmias, labile BP (50-70%) |
| Bowel Care |
Stool softeners, schedule |
Grade C |
Ileus, constipation |
| Physical Therapy |
Passive ROM, early mobilization when stable |
Grade B |
Prevent contractures |
| Nutritional Support |
NG/PEG if dysphagia, high protein |
Grade B |
Aspiration risk if bulbar weakness |
Treatment Algorithm:
Phase 1: Diagnosis & Admission (Day 0-1) 1. Confirm diagnosis clinically + CSF + EMG/NCS [Grade A] 2. Admit to ICU/monitored bed if any of: FVC <40 mL/kg, bulbar weakness, autonomic instability, rapid progression [Grade A] 3. Baseline respiratory function: FVC, NIF q4-6h [Grade A]
Phase 2: Immunotherapy (Day 1-2, within 2 weeks of onset) 1. First-line: Choose IVIG OR PLEX (do NOT combine—no added benefit) [Grade A] - IVIG 0.4 g/kg/day × 5 days (preferred if autonomic instability) - PLEX 4-6 sessions (if IVIG contraindicated or very severe)
- Do NOT use corticosteroids [Grade D—ineffective]
Phase 3: Supportive Care (Throughout hospitalization) 1. Respiratory: - Monitor FVC/NIF q4-6h - Intubate if FVC <20 mL/kg, NIF >-30 cmH₂O, or respiratory distress [Grade A]
- Autonomic:
- Continuous telemetry
- Treat bradycardia (atropine), tachycardia (short-acting β-blockers), labile BP (avoid aggressive treatment)
- Prophylaxis:
- DVT prophylaxis (enoxaparin/heparin) [Grade A]
- Pain control (gabapentin, carbamazepine) [Grade B]
- Bowel/bladder care
Phase 4: Plateau & Early Recovery (Weeks 2-4) 1. Monitor for complications: Hospital-acquired infections, pressure ulcers, contractures 2. Early mobilization when stable [Grade B] 3. Continue physical/occupational therapy
Phase 5: Rehabilitation (Months 1-6+) 1. Inpatient rehab if significant weakness persists 2. Outpatient therapy for residual deficits 3. Monitor for CIDP (chronic form)—5-10% may develop chronic relapsing course
Prognosis: - 70-80% achieve full or near-full recovery [Grade A evidence] - 15-20% have residual weakness or fatigue - 3-5% mortality (respiratory failure, autonomic complications, PE) - Recovery timeline: Plateau at 2-4 weeks, improvement over 6-12 months - Poor prognostic factors: Age >60, rapid progression, axonal involvement on EMG, need for mechanical ventilation
Multiple Choice Question
Question: Which of the following findings is most characteristic of AIDP?
- Rapidly ascending symmetric weakness with hyperreflexia
- Progressive weakness with areflexia and albuminocytologic
dissociation in CSF (C) Fluctuating weakness with preserved reflexes and fatigability (D) Sensory ataxia with preserved motor strength
Answer: (B) Progressive weakness with areflexia and albuminocytologic dissociation in CSF
Cervical Spondylosis with Chronic Radiculopathy
📊 Quick Facts
- Prevalence: Radiographic spondylosis in >85% of adults >60 years
- Peak Age: 50–70 years; radiculopathy peaks at 40–50 years
- Gender: M > F (approximately 1.5:1)
- Most Affected Levels: C5–C6, C6–C7 (>70% of cases)
- Chronicity: Symptoms persisting >12 weeks define chronic radiculopathy
⚠️ Red Flags
- Progressive myelopathic signs → Urgent surgical evaluation
- Bilateral upper extremity weakness → Rule out cord compression
- Bowel/bladder dysfunction → Consider cauda equina or myelopathy
- Rapid onset in young patient → Disc herniation, tumor, or infection
- Weight loss, fever, night pain → Malignancy or epidural abscess
Clinical Scenario
A 55-year-old right-handed man presents with a 6-month history of persistent right-sided neck pain radiating into the lateral forearm and thumb.
He reports intermittent numbness and tingling in the right thumb and index finger that worsen with neck extension and right lateral rotation.
On examination, there is diminished biceps reflex on the right, weakness of elbow flexion and wrist extension (4/5), and decreased sensation in the C6 dermatome.
Spurling’s test reproduces the radicular pain on the right side.
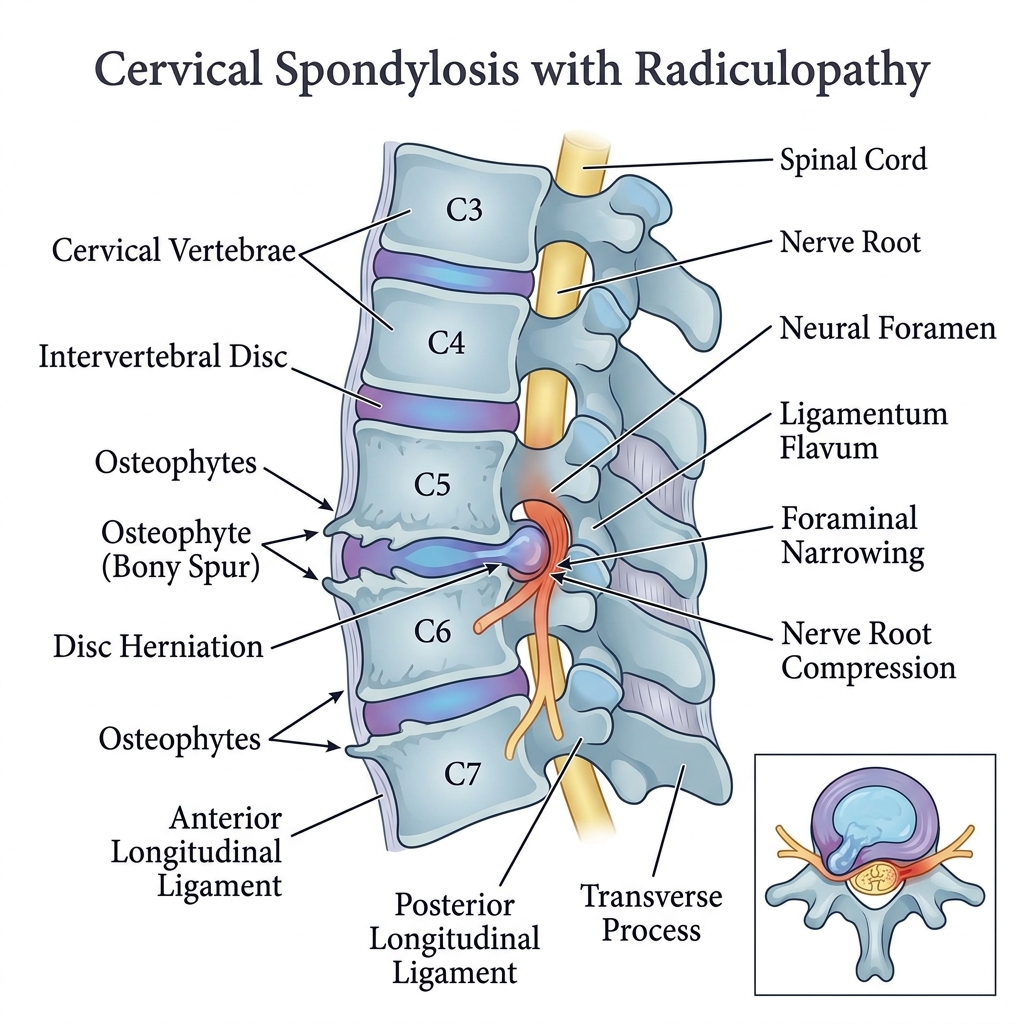
MRI of the cervical spine reveals C5–C6 disc-osteophyte complex with right-sided foraminal stenosis and nerve root compression.
Epidemiology
Cervical spondylosis is the most common cause of cervical radiculopathy, accounting for approximately 70–75% of cases, with disc herniation responsible for most of the remainder.
Radiographic evidence of cervical spondylosis is present in more than 85% of individuals over 60 years, though only a minority develop symptomatic radiculopathy.
The annual incidence of cervical radiculopathy is approximately 83 per 100,000 population, with a peak incidence in the fifth decade.
Males are affected slightly more frequently, with a male-to-female ratio of approximately 1.5:1.
Risk factors include advancing age, repetitive axial loading, prior cervical injury, smoking, and occupations involving heavy manual labor or prolonged neck flexion.
Etiopathophysiology
- Cervical spondylosis involves progressive degeneration of the intervertebral discs, uncovertebral joints, and facet joints, leading to osteophyte formation and loss of disc height.
- Disc desiccation and annular weakening result in posterior or posterolateral disc bulging that encroaches on the neural foramina.
- Osteophytic overgrowth from the uncovertebral joints (joints of Luschka) further narrows the foramina, compressing the exiting nerve roots.
- Chronic nerve root compression leads to demyelination, axonal injury, intraneural edema, and ischemia from impaired vascular supply to the nerve root.
- Inflammatory mediators (e.g., TNF-α, IL-6, substance P) released from the degenerative disc contribute to neuropathic pain even without direct mechanical compression.
- The transition from acute to chronic radiculopathy (>12 weeks) involves central sensitization, dorsal horn neuroplastic changes, and peripheral nerve fibrosis.
Clinical Features
The hallmark presentation is unilateral neck pain radiating into the arm along a dermatomal distribution, most commonly C6 (lateral forearm, thumb) or C7 (posterior forearm, middle finger).
Patients describe burning, electric, or lancinating pain that may be accompanied by numbness, tingling, and paresthesias in the affected dermatome.
Motor weakness corresponding to the affected root is common: C5 (deltoid, biceps), C6 (wrist extensors, biceps), C7 (triceps, wrist flexors, finger extensors), C8 (finger flexors, hand intrinsics).
Diminished deep tendon reflexes in the corresponding myotome are characteristic: biceps (C5–C6), brachioradialis (C5–C6), triceps (C7).
Chronic cases may demonstrate muscle atrophy in the affected myotome, persistent neuropathic pain features (allodynia, hyperalgesia), and reduced functional capacity.
Spurling’s test (cervical extension + ipsilateral rotation + axial compression) has high specificity (~95%) but modest sensitivity (~50%) for cervical radiculopathy. A positive test strongly supports the diagnosis, but a negative test does not exclude it. Combine with shoulder abduction relief sign (Bakody’s sign) to improve diagnostic yield.
Diagnosis and Differential Diagnosis
MRI of the cervical spine is the gold standard, demonstrating disc-osteophyte complexes, foraminal stenosis, and nerve root compression with high sensitivity and specificity.
Electrodiagnostic studies (EMG/NCS) are valuable in chronic cases to confirm radiculopathy, exclude peripheral nerve entrapment (e.g., carpal tunnel syndrome), assess severity of axonal loss, and guide prognosis.
CT myelography is an alternative when MRI is contraindicated and provides excellent visualization of bony foraminal stenosis.
Plain radiographs may show loss of disc height, osteophytes, and foraminal narrowing but lack sensitivity for soft tissue pathology.
Differential diagnoses include cervical myelopathy, brachial plexopathy (Parsonage-Turner syndrome), peripheral nerve entrapments (carpal tunnel, cubital tunnel), thoracic outlet syndrome, rotator cuff pathology, and inflammatory or neoplastic radiculopathy.
Cervical Root Localization Guide:
| C5 |
C4–C5 |
Lateral shoulder, upper arm |
Deltoid, biceps |
Biceps |
| C6 |
C5–C6 |
Lateral forearm, thumb, index finger |
Wrist extensors, biceps |
Brachioradialis |
| C7 |
C6–C7 |
Posterior forearm, middle finger |
Triceps, wrist flexors, finger extensors |
Triceps |
| C8 |
C7–T1 |
Medial forearm, ring/little finger |
Finger flexors, hand intrinsics |
None |
| T1 |
T1–T2 |
Medial upper arm |
Hand intrinsics |
None |
- MRI cervical spine (with or without contrast): First-line imaging to confirm root compression and exclude cord compression, tumor, or infection.
- EMG/NCS (if >3–4 weeks of symptoms): Confirms radiculopathy, localizes affected root, differentiates from plexopathy or peripheral neuropathy, and assesses chronicity.
- Plain radiographs (AP, lateral, oblique): Baseline assessment of alignment, disc height, and osteophytes.
- CT myelography: If MRI contraindicated; superior for foraminal bony stenosis detail.
- Laboratory studies: ESR, CRP, CBC if infection or malignancy suspected; B12, HbA1c if concurrent neuropathy considered.
Management
Conservative Management (First-line for most chronic cases):
| Physical Therapy |
Cervical traction, isometric strengthening, postural correction, nerve gliding exercises; evidence supports 6–12 weeks of structured PT |
| Pharmacotherapy |
NSAIDs (first-line), gabapentin/pregabalin for neuropathic pain, short-course muscle relaxants, duloxetine for chronic pain |
| Cervical Epidural Steroid Injections |
Transforaminal or interlaminar approach; provides 50–75% short-to-intermediate relief; useful as bridge to surgery or adjunct to PT |
| Activity Modification |
Ergonomic optimization, avoidance of sustained neck extension, cervical pillow for sleep positioning |
| Cervical Collar |
Short-term use (1–2 weeks) for acute flares; avoid prolonged use to prevent deconditioning |
Surgical Management (for refractory or progressive cases):
| Single-level disc herniation |
Anterior cervical discectomy and fusion (ACDF) |
Gold standard; >90% success rate |
| Multilevel spondylotic stenosis |
Anterior corpectomy or posterior laminectomy ± fusion |
For 3+ levels with significant stenosis |
| Foraminal stenosis (bony) |
Posterior foraminotomy |
Preserves motion; suitable for lateral soft disc or osteophyte |
| Failed conservative therapy >6–12 weeks |
Surgical evaluation |
Persistent motor deficit, intractable pain, or progressive weakness |
Management Algorithm: 1. Initial presentation: Confirm diagnosis with MRI ± EMG; initiate conservative therapy (NSAIDs, PT, activity modification). 2. 4–6 weeks: Reassess; if inadequate response, add neuropathic pain agents (gabapentin/pregabalin), consider epidural steroid injection. 3. 6–12 weeks: If refractory, repeat imaging, advanced electrodiagnostics; surgical consultation for progressive weakness, intractable pain, or functional decline. 4. Surgical candidates: ACDF for single/two-level disease; posterior approach for multilevel or lateral pathology; postoperative rehabilitation for 6–12 weeks. 5. Long-term: Maintain cervical strengthening, ergonomic optimization, and periodic reassessment for adjacent segment disease.
Multiple Choice Question
A 52-year-old man presents with 4 months of right arm pain radiating to the thumb and index finger, with weakness of wrist extension. Spurling’s test is positive on the right. MRI shows a C5–C6 disc-osteophyte complex with right foraminal stenosis. Which nerve root is most likely affected?
- C5
- C6
- C7
- C8
- T1
Answer: (B) C6 radiculopathy presents with pain radiating to the lateral forearm, thumb, and index finger, weakness of wrist extensors (and biceps), and diminished brachioradialis reflex. The C5–C6 disc level compresses the exiting C6 nerve root.
References
- Iyer S, Kim HJ. Cervical radiculopathy. Curr Rev Musculoskelet Med. 2016;9(3):272–280. DOI: 10.1007/s12178-016-9349-4
- Woods BI, Hilibrand AS. Cervical radiculopathy: epidemiology, etiology, diagnosis, and treatment. J Spinal Disord Tech. 2015;28(5):E251–E259. DOI: 10.1097/BSD.0000000000000284
- Caridi JM, Pumberger M, Hughes AP. Cervical radiculopathy: a review. HSS J. 2011;7(3):265–272. DOI: 10.1007/s11420-011-9218-z
- Bono CM, Ghiselli G, Gilbert TJ, et al. An evidence-based clinical guideline for the diagnosis and treatment of cervical radiculopathy from degenerative disorders. Spine J. 2011;11(1):64–72. DOI: 10.1016/j.spinee.2010.10.023
- Thoomes EJ, Scholten-Peeters W, Windt DA, et al. The effectiveness of conservative treatment for patients with cervical radiculopathy: a systematic review. Clin J Pain. 2013;29(12):1073–1086. DOI: 10.1097/AJP.0b013e31828441fb
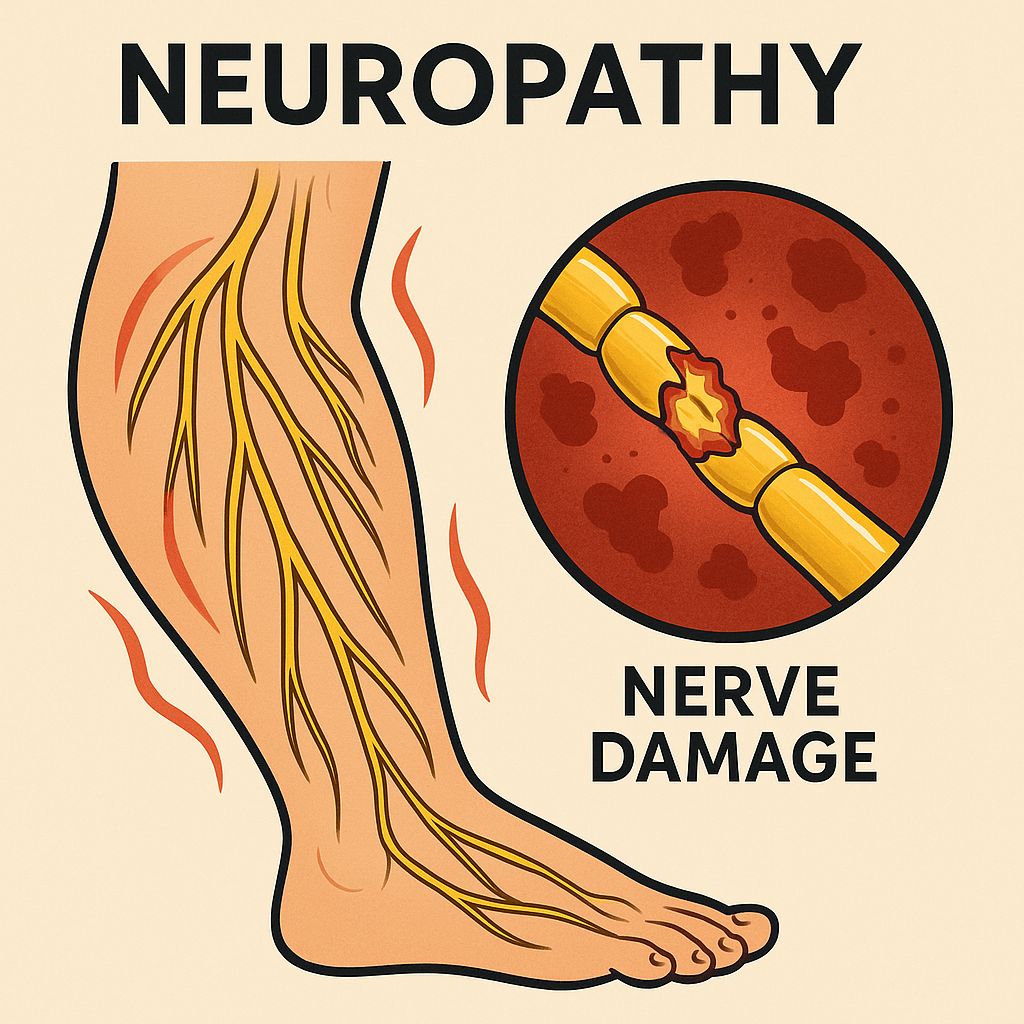
Diabetic Neuropathy
📊 Quick Facts
- Prevalence: Up to 50% of long-standing diabetes
- Patterns: Distal symmetric sensorimotor polyneuropathy most common; also autonomic, focal, and proximal forms
- Risk Factors: Duration, poor glycemic control, hypertension, dyslipidemia, smoking
- Complications: Foot ulcers, Charcot arthropathy, falls
- Prevention: Intensive glycemic control especially in type 1 diabetes
⚠️ Red Flags
- Rapid progression, asymmetry, or proximal weakness → evaluate for CIDP or vasculitis
- Motor predominance with weight loss → diabetic radiculoplexus neuropathy (amyotrophy)
- Severe autonomic failure → check for autoimmune autonomic ganglionopathy
- Acute cranial neuropathy → rule out stroke or compressive lesions
- Normal glycemic control but neuropathy → reassess diagnosis (B12 deficiency, toxins)
Clinical Scenario
A 58-year-old man with a 15-year history of poorly controlled type 2 diabetes mellitus presents with burning pain and numbness in his feet, which has gradually progressed over the past year.
He reports difficulty walking in the dark and frequent falls due to imbalance.
Examination reveals decreased vibration and pinprick sensation in a stocking distribution, absent ankle reflexes, and mild distal muscle weakness.
There is no evidence of acute foot ulceration or infection, but he has reduced proprioception in his toes.
Autonomic symptoms include orthostatic hypotension and erectile dysfunction.
Epidemiology
Diabetic neuropathy affects up to 50% of patients with long-standing diabetes mellitus.
It is the most common cause of peripheral neuropathy in developed countries.
The risk increases with duration of diabetes, poor glycemic control, and presence of comorbidities like hypertension and dyslipidemia.
Both type 1 and type 2 diabetes can lead to neuropathy, though it is more prevalent in type 2 due to longer undiagnosed disease duration.
Peripheral symmetric polyneuropathy is the most frequent form, but focal and autonomic neuropathies also occur.
Etiopathophysiology
- Chronic hyperglycemia activates the polyol pathway (sorbitol accumulation), increases oxidative stress, and promotes advanced glycation end products that damage neurons and Schwann cells.
- Microvascular ischemia from endothelial dysfunction compromises nutrient delivery to nerves.
- Inflammatory mediators and impaired neurotrophic signaling accelerate axonal degeneration and demyelination.
- Autonomic fibers suffer similar metabolic/ischemic insults, causing cardiovascular and GI dysautonomia.
- Genetic susceptibility, obesity, and smoking worsen oxidative injury and microvascular disease.
Clinical Features
Sensory symptoms include burning, tingling, numbness, and lancinating pain, typically in a symmetric distal pattern ("stocking-glove" distribution).
Motor involvement may manifest as distal weakness, muscle wasting, and gait instability.
Autonomic features include orthostatic hypotension, gastrointestinal dysmotility, bladder dysfunction, and sexual dysfunction.
Diabetic amyotrophy presents with painful asymmetric proximal weakness, often in the thighs.
Mononeuropathies, including cranial nerve palsies, can occur acutely and resolve spontaneously.
“Stocking-glove” sensory loss with absent ankle reflexes and preserved proprioception above the ankle strongly suggests diabetic peripheral neuropathy; evaluate for autonomic involvement at each visit (orthostatic vitals, gastrointestinal symptoms).
Diagnosis and Differential Diagnosis
Diagnosis is primarily clinical, supported by nerve conduction studies showing distal symmetric sensorimotor polyneuropathy.
Laboratory workup includes HbA1c, B12 levels, thyroid function tests, and serum protein electrophoresis to exclude other causes.
Differential diagnoses include chronic inflammatory demyelinating polyneuropathy (CIDP), vitamin deficiencies, alcohol-related neuropathy, and paraproteinemic neuropathy.
Skin biopsy or corneal confocal microscopy may aid in diagnosing small fiber neuropathy.
Quantitative sensory testing and autonomic function testing can provide further diagnostic clarity.
Differential Diagnosis Comparison:
| Onset |
Gradual, chronic |
Subacute/chronic (>8 wks) |
Gradual |
Gradual |
| Pattern |
Distal symmetric |
Proximal + distal, motor > sensory |
Sensory ataxia, posterior column signs |
Distal symmetric |
| Reflexes |
Reduced distally |
Globally reduced |
Reduced Achilles, preserved knees early |
Reduced |
| NCS/EMG |
Axonal loss |
Demyelinating features |
Axonal sensory > motor |
Axonal loss |
| Labs |
Elevated HbA1c |
Elevated CSF protein |
Low B12, elevated MMA |
Elevated liver enzymes |
| Treatment |
Glycemic + risk-factor control |
Immunotherapy |
B12 replacement |
Abstinence, nutrition |
- Clinical pattern: Distal symmetric symptoms, motor involvement, autonomic complaints.
- Laboratory evaluation: HbA1c, B12, TSH, renal function, SPEP to exclude other causes.
- Neurophysiology: NCS/EMG for atypical features or rapid progression.
- Autonomic testing: Tilt-table, QSART when autonomic symptoms predominate.
- Screen for complications: Foot exam, ABI, evaluation for Charcot arthropathy and retinopathy.
Management
Disease-Modifying Strategies:
| Glycemic control |
Intensive control reduces incidence (especially in type 1); avoid hypoglycemia |
| Cardiometabolic optimization |
Manage hypertension, dyslipidemia, obesity, smoking |
| Lifestyle |
Exercise, weight management, smoking cessation |
Neuropathic Pain Control:
| Gabapentinoids |
Gabapentin, pregabalin |
Grade A |
First-line; titrate slowly |
| SNRIs |
Duloxetine, venlafaxine |
Grade A |
Beneficial for comorbid mood disorders |
| TCAs |
Amitriptyline, nortriptyline |
Grade B |
Use lower doses in elderly |
| Topicals |
Capsaicin 8% patch, lidocaine 5% patch |
Grade B |
For focal pain |
| Opioids/tramadol |
Reserved for refractory cases |
Grade C |
Short-term use only |
Autonomic Dysfunction Management: - Orthostatic hypotension: increased fluids/salt, compression stockings, midodrine/fludrocortisone. - Gastroparesis: dietary changes, prokinetics (metoclopramide, erythromycin). - Bladder dysfunction: timed voiding, intermittent catheterization; consider bethanechol. - Erectile dysfunction: PDE-5 inhibitors; address cardiovascular risk.
Supportive Care: - Foot care education, podiatry follow-up, custom footwear. - Physical therapy for balance training and fall prevention. - Psychological support for chronic pain and quality-of-life issues.
Management Algorithm: 1. Assess pain, motor, and autonomic involvement at each visit. 2. Optimize glycemic and cardiometabolic control; counsel on lifestyle changes. 3. Initiate pain therapy (gabapentinoid or SNRI) with stepwise titration; add topical agents for focal pain. 4. Treat autonomic symptoms with targeted pharmacologic and nonpharmacologic measures. 5. Prevent complications with regular foot exams, patient education, and early referral to podiatry/rehab services.
Multiple Choice Question
Question: Which of the following is the most common clinical presentation of diabetic neuropathy?
- Acute mononeuropathy
- Proximal motor neuropathy
- Symmetric distal sensorimotor polyneuropathy
- Cranial nerve palsy
Answer: (C) Symmetric distal sensorimotor polyneuropathy
References
- Tesfaye S, Boulton AJM, Dyck PJ, et al. Diabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care. 2010;33(10):2285–2293. https://doi.org/10.2337/dc10-1303
- Pop-Busui R, Boulton AJ, Feldman EL, et al. Diabetic neuropathy: a position statement by the American Diabetes Association. Diabetes Care. 2017;40(1):136–154. https://doi.org/10.2337/dc16-2042
Inclusion Body Myositis

📊 Quick Facts
- Prevalence: 2-7 per 100,000
- Peak Age: 55-65 years
- Gender: M:F = 2-3:1
- Course: Slowly progressive
⚠️ Red Flags
- Asymmetric finger flexor + quadriceps weakness = Hallmark
- Normal/mild CK (contrast to polymyositis)
- Refractory to steroids/immunosuppression
- Progressive course despite treatment
Clinical Scenario
A 64-year-old man presents with progressive difficulty climbing stairs and frequent falls over the past 3 years.
He also reports trouble gripping objects and dropping utensils due to hand weakness.
Neurological examination reveals asymmetric quadriceps and finger flexor weakness, with preserved sensation.
Reflexes are normal or slightly reduced, and there is no muscle pain.
Laboratory testing shows mildly elevated creatine kinase (CK), and EMG reveals a myopathic pattern.
Epidemiology
Inclusion Body Myositis is the most common idiopathic inflammatory myopathy in adults over 50 years.
The prevalence is estimated at 2–7 per 100,000, with a male predominance (male:female ≈ 2:1 to 3:1).
Mean age of onset is typically 55–65 years, rarely occurring before age 45.
Disease onset is insidious, often leading to diagnostic delays exceeding 3–5 years (average diagnostic delay: 5–7 years).
IBM is usually sporadic (sIBM), though rare familial/hereditary cases (hIBM) have been reported with earlier onset and recessive inheritance.
The condition progresses slowly but relentlessly, leading to significant disability, with most patients requiring assistive devices within 10–15 years.
Higher prevalence in Caucasian populations compared to other ethnic groups.
Etiopathophysiology
IBM is characterized by a unique combination of inflammatory and degenerative mechanisms affecting skeletal muscle, distinguishing it from other inflammatory myopathies.
Inflammatory Component:
- Cytotoxic CD8+ T-cell infiltration targets muscle fibers expressing MHC class I molecules, causing endomysial inflammation
- Upregulation of MHC-I on muscle fiber sarcolemma is characteristic
- T-cell mediated cytotoxicity results in non-necrotic muscle fiber invasion
Degenerative Component:
- Intracellular protein aggregates, including amyloid-β (Aβ), phosphorylated tau, TDP-43, and α-synuclein accumulate within muscle fibers—resembling neurodegenerative diseases
- Rimmed vacuoles contain accumulated proteins and cellular debris
- Mitochondrial abnormalities include cytochrome oxidase (COX)-negative fibers and ragged-red fibers
- Impaired autophagy and proteasomal degradation contribute to progressive fiber degeneration
Genetic Susceptibility:
- HLA-DR3 allele is strongly associated with IBM susceptibility
- Polymorphisms in genes related to immunity and protein degradation may increase risk
Key Mechanisms:
- Dual pathology: Immune-mediated inflammation + protein aggregation
- Molecular mimicry between self-antigens and muscle proteins
- Mitochondrial dysfunction and oxidative stress
- Impaired protein degradation pathways (autophagy, proteasome)
- The interplay between immune-mediated injury, protein misfolding, and mitochondrial dysfunction underlies the chronic, treatment-resistant course
Clinical Features
Progressive, asymmetric muscle weakness is the hallmark—a key distinguishing feature from polymyositis/dermatomyositis.
Quadriceps weakness (knee extensors): Leading cause of falls, difficulty rising from chairs, and stair climbing.
Deep finger flexor weakness (FDP > FDS): Difficulty with grip, turning keys, opening jars; “finger flexor sign” is characteristic.
Ankle dorsiflexors: Foot drop may develop, contributing to falls.
Proximal muscles (deltoids, hip flexors) are also affected but typically later in disease.
Dysphagia occurs in 40–60% of patients due to involvement of pharyngeal muscles; risk of aspiration pneumonia.
Muscle atrophy becomes prominent as disease progresses, particularly in the forearms and thighs.
Unlike polymyositis or dermatomyositis, IBM has a slow, indolent course (months to years) and often presents with falls or grip weakness rather than acute proximal weakness.
Sensory function is completely preserved—a critical diagnostic clue.
Muscle pain is uncommon (contrast with polymyositis).
Reflexes may be diminished or absent in severely affected muscles.
No skin rash (unlike dermatomyositis).
Muscle wasting becomes evident as the disease advances, leading to significant functional impairment and wheelchair dependence in 10–20 years.
Early-onset IBM: Rare, often hereditary, presenting before age 40.
IBM with systemic autoimmune disease: Coexistence with Sjögren’s syndrome, SLE, or sarcoidosis in 15–20% of cases.
Diagnosis
Serum CK levels are mildly elevated or normal (often 2–10× normal)—much lower than polymyositis (which can be 10–50× normal).
Anti-cN1A (cytosolic 5’-nucleotidase 1A) antibodies present in 30–40% of IBM patients (more specific but not diagnostic).
Electromyography reveals a mixed myopathic-neurogenic pattern with spontaneous activity (fibrillations, positive sharp waves).
MRI shows selective involvement of quadriceps (especially vastus medialis and lateralis), finger flexors, and ankle dorsiflexors with fatty replacement and atrophy.
Muscle biopsy is diagnostic, showing endomysial inflammation, rimmed vacuoles, and intracellular inclusions (amyloid-β, TDP-43, p62).
Pathological hallmarks (requires all three for definite diagnosis): (1) CD8+ T-cell endomysial inflammation, (2) rimmed vacuoles, (3) protein accumulation.
Additional biopsy findings: Increased MHC-I expression, COX-negative fibers, tubulofilamentous inclusions on electron microscopy.
Differential diagnoses include polymyositis, ALS, limb-girdle muscular dystrophy, myasthenia gravis, and hereditary inclusion body myopathies.
Genetic testing (VCP, GNE, MYH7, MATR3 genes) may be performed to exclude hereditary inclusion body myopathies when the presentation is atypical.
Management
IBM is generally refractory to immunosuppressive therapies—a key distinguishing feature from polymyositis/dermatomyositis.
Corticosteroids are typically ineffective and may worsen weakness; generally not recommended.
IVIG may provide modest, temporary improvement in 10–30% of patients, but no sustained benefit in controlled trials.
Methotrexate, azathioprine, and mycophenolate are generally ineffective; not routinely recommended.
Physical therapy with strengthening exercises, range-of-motion training, and gait training helps preserve function and prevent contractures.
Occupational therapy provides adaptive devices (button hooks, jar openers, zipper pulls) and assistive technology for activities of daily living.
Ankle-foot orthoses (AFOs) may be needed for foot drop; mobility aids (cane, walker, wheelchair) as disease progresses.
Dysphagia management includes dietary modification, swallowing techniques, and gastrostomy tube (PEG) placement in severe cases.
Regular neuromuscular clinic visits every 3–6 months for strength assessments, swallowing evaluation, and screening for aspiration pneumonia.
Prognosis: Slowly progressive disease with median time to wheelchair dependence of 10–15 years; life expectancy often near normal.
No curative treatment currently available; management is entirely supportive and symptomatic.
Multiple Choice Question
Which of the following is most characteristic of inclusion body myositis?
- Rapidly progressive symmetric proximal muscle weakness
- Presence of anti-Mi-2 antibodies
- Asymmetric involvement of quadriceps and finger flexors with rimmed vacuoles on biopsy
- Dramatic improvement with corticosteroid therapy
Answer: (C) Asymmetric involvement of quadriceps and finger flexors with rimmed vacuoles on biopsy is the hallmark of IBM. Unlike other inflammatory myopathies, IBM presents with asymmetric distal and proximal weakness, is refractory to immunosuppression, and requires all three pathological features (inflammation, rimmed vacuoles, protein inclusions) for definite diagnosis.
References
Needham M, Mastaglia FL. Inclusion body myositis: current pathogenetic concepts and diagnostic and therapeutic approaches. Lancet Neurol. 2007;6(7):620–631. https://doi.org/10.1016/S1474-4422(07)70171-0
Dimachkie MM, Barohn RJ. Inclusion body myositis. Neurol Clin. 2014;32(3):629–646. https://doi.org/10.1016/j.ncl.2014.04.007
Griggs RC, Askanas V, DiMauro S, et al. Inclusion body myositis and myopathies. Ann Neurol. 1995;38(5):705–713. https://doi.org/10.1002/ana.410380504
Rose MR, ENMC IBM Working Group. 188th ENMC International Workshop: Inclusion Body Myositis, 2-4 December 2011, Naarden, The Netherlands. Neuromuscul Disord. 2013;23(12):1044–1055. https://doi.org/10.1016/j.nmd.2013.08.007
Benveniste O, Guiguet M, Freebody J, et al. Long-term observational study of sporadic inclusion body myositis. Brain. 2011;134(11):3176–3184. https://doi.org/10.1093/brain/awr213
Lewy Body Dementia (LBD)

📊 Quick Facts
- Prevalence: 10-15% of degenerative dementias
- Peak Age: 65-75 years
- Gender: Slight male predominance
- Pathology: Cortical α-synuclein + frequent Alzheimer co-pathology
- Course: Cognitive decline within 1 year of parkinsonism
⚠️ Red Flags
- Severe neuroleptic sensitivity → Think LBD, avoid typical antipsychotics
- Early complex visual hallucinations → LBD over AD
- REM sleep behavior disorder years before dementia → Prodromal LBD
- Pronounced fluctuations in alertness → Suggests LBD
- Autonomic failure (orthostasis, constipation) → Supports diagnosis
Clinical Scenario
A 72-year-old man presents with progressive cognitive decline over one year, marked by fluctuating attention and recurrent visual hallucinations.
His wife describes vivid, well-formed hallucinations of people and animals, alongside episodes of confusion lasting several hours.
Neurological examination reveals mild bradykinesia and rigidity without tremor, and no history of antipsychotic use.
Cognitive testing shows impaired visuospatial and executive function with relatively preserved memory.
These findings are consistent with Lewy body dementia (LBD), a neurodegenerative disorder combining features of Alzheimer’s and Parkinson’s disease.
Epidemiology
LBD is the second most common cause of degenerative dementia after Alzheimer’s disease, responsible for 10–15% of cases.
The average age of onset is 65–75 years, with a slight male predominance.
Prevalence rises with age, affecting up to 5% of individuals older than 85 years.
LBD shares clinical overlap with Parkinson’s disease dementia (PDD), but cognitive impairment appears first or within one year of motor symptoms.
Risk factors include advanced age, REM sleep behavior disorder (RBD), and genetic variants such as GBA mutations.
Etiopathophysiology
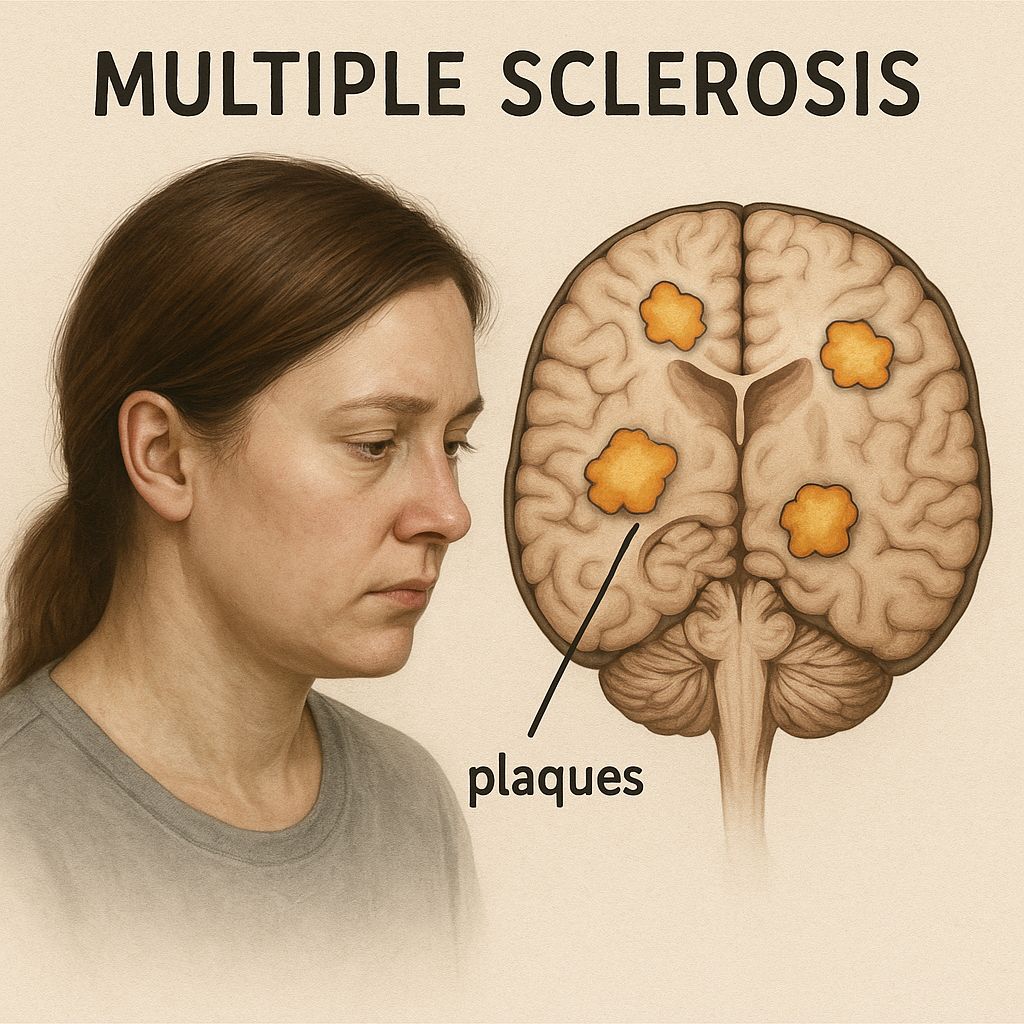
LBD is caused by accumulation of misfolded α-synuclein forming Lewy bodies and neurites within cortical and limbic neurons.
These inclusions disrupt synaptic transmission, mitochondrial function, and axonal transport, producing widespread network dysfunction.
Loss of cholinergic neurons in the basal forebrain contributes to visual hallucinations and attention deficits, while nigrostriatal dopaminergic loss causes parkinsonism.
Concomitant Alzheimer pathology (amyloid-β plaques, tau tangles) is frequent and accelerates cognitive decline.
Prodromal REM sleep behavior disorder reflects early brainstem involvement before cortical spread.
Clinical Features
Core features include fluctuating cognition, recurrent visual hallucinations**, and spontaneous parkinsonism.
REM sleep behavior disorder (RBD), characterized by dream enactment, may precede cognitive symptoms by several years.
Patients show marked sensitivity to antipsychotics, leading to severe extrapyramidal reactions.
Cognitive dysfunction primarily affects attention, visuospatial ability, and executive function, with early memory sparing.
Non-motor features include autonomic dysfunction, mood changes, and delusions.
Use the “central triad”: fluctuations + visual hallucinations + parkinsonism within 12 months. Severe worsened rigidity after standard antipsychotics is nearly pathognomonic—switch to quetiapine, clozapine, or pimavanserin.
Diagnosis and Differential Diagnosis
Diagnosis is primarily clinical, guided by consensus criteria from the DLB Consortium.
Neuroimaging may reveal occipital hypometabolism on FDG-PET or reduced dopamine transporter uptake on DaT-SPECT.
Polysomnography confirms RBD, and MIBG cardiac scintigraphy often shows reduced sympathetic innervation.
Key differentials include Alzheimer’s disease, PDD, vascular dementia, and normal pressure hydrocephalus.
The distinction from PDD lies in timing: in LBD, cognitive impairment precedes or occurs within one year of parkinsonism onset.
Differential Diagnosis Comparison:
| Onset |
Dementia + parkinsonism within 1 yr |
Gradual memory-first decline |
Parkinsonism ≥1 yr before dementia |
Stepwise |
| Hallucinations |
Early, complex visual |
Late, less formed |
Late |
Rare |
| Fluctuations |
Prominent |
Mild |
Mild |
Variable |
| REM Sleep Behavior Disorder |
Common, prodromal |
Uncommon |
Common |
Rare |
| Imaging |
Occipital hypometabolism, DaTscan ↓ |
Hippocampal atrophy |
DaTscan ↓ |
White matter disease |
| Drug Sensitivity |
Severe to typical antipsychotics |
Mild |
Moderate |
Mild |
Diagnosis of probable LBD requires: 1. Dementia plus ≥2 core clinical features (fluctuations, recurrent visual hallucinations, spontaneous parkinsonism, REM sleep behavior disorder) OR 2. Dementia plus 1 core feature AND ≥1 indicative biomarker, such as: - DaT-SPECT showing reduced striatal uptake - Polysomnography-confirmed REM sleep without atonia - MIBG scintigraphy showing low cardiac uptake
Possible LBD: Dementia + 1 core feature, or dementia + indicative biomarker without core features. Exclude alternative causes (e.g., stroke, medication effects).
Management
Pharmacologic Therapy:
| Cognition / Hallucinations |
Rivastigmine (PO or patch) |
6-12 mg/day PO or 9.5 mg patch |
Grade A |
Improves attention, hallucinations |
|
Donepezil |
5-10 mg/day |
Grade A |
Similar cognitive benefit |
| Motor symptoms |
Levodopa/carbidopa |
Lowest effective dose |
Grade B |
May worsen psychosis; titrate cautiously |
| Psychosis |
Quetiapine 12.5-50 mg HS |
Grade B |
Use if distressing hallucinations; monitor QT |
|
|
Clozapine 12.5-25 mg/day |
Grade B |
Requires ANC monitoring; least motor worsening |
|
|
Pimavanserin 34 mg/day |
Grade B |
Approved for PD psychosis; helpful in LBD |
|
| REM Sleep Behavior Disorder |
Melatonin 3-12 mg HS |
Grade B |
First-line, minimal side effects |
|
|
Clonazepam 0.25-1 mg HS |
Grade C |
Use if melatonin insufficient |
|
| Autonomic Dysfunction |
Midodrine, fludrocortisone, droxidopa |
Standard doses |
Grade C |
Treat orthostasis/constipation individually |
Management Algorithm:
- Diagnosis & Education: Explain 1-year rule, neuroleptic sensitivity; involve caregivers early.
- First-line therapy: Start rivastigmine or donepezil; address REM sleep behavior disorder (melatonin).
- Motor symptoms: Trial low-dose levodopa if disabling parkinsonism; reassess hallucinations after dose change.
- Psychosis/Behavior: If hallucinations distressing, use quetiapine or pimavanserin; avoid typical antipsychotics (haloperidol).
- Autonomic & Sleep: Manage orthostatic hypotension, constipation, urinary symptoms; treat insomnia/RBD.
- Supportive Care: Physical/occupational therapy for gait instability, speech therapy for hypophonia, caregiver respite, advance care planning.
Multiple Choice Question
Which of the following features most strongly supports a diagnosis of Lewy body dementia over Alzheimer’s disease?
- Early memory loss with hippocampal atrophy on MRI
- Fluctuating cognition, recurrent visual hallucinations, and
- parkinsonism occurring within one year of each other
- Gradual visuospatial decline without hallucinations
- Stepwise progression of cognitive deficits
Answer: (B) Fluctuating cognition, recurrent visual hallucinations, and arkinsonism occurring within one year of each other are classic features of Lewy body dementia.
References
McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology. 2017;89(1):88–100. DOI: 10.1212/WNL.0000000000004058
Walker Z, Possin KL, Boeve BF, Aarsland D. Lewy body dementias. Lancet. 2015;386(10004):1683–1697. DOI: 10.1016/S0140-6736(15)00462-6
Taylor JP, McKeith IG, Burn DJ, et al. New evidence on the management of Lewy body dementia. Lancet Neurol. 2020;19(2):157–169. DOI: 10.1016/S1474-4422(19)30153-X
Lumbar Spondylosis with Chronic Radiculopathy
📊 Quick Facts
- Prevalence: Radiographic lumbar spondylosis in >90% of adults >50 years
- Peak Age: 40–60 years for symptomatic radiculopathy
- Gender: M ≈ F (slight male predominance in surgical cases)
- Most Affected Levels: L4–L5 and L5–S1 (>95% of lumbar radiculopathies)
- Chronicity: Symptoms persisting >12 weeks; 20–30% of acute cases progress to chronic
⚠️ Red Flags
- Saddle anesthesia + urinary retention → Cauda equina syndrome (surgical emergency)
- Progressive bilateral leg weakness → Central disc herniation or spinal stenosis
- Night pain, weight loss, fever → Malignancy or epidural abscess
- Trauma in osteoporotic patient → Pathologic fracture
- Rapidly progressive foot drop → Urgent decompression
Clinical Scenario
A 58-year-old construction worker presents with an 8-month history of persistent low back pain radiating down the left posterior thigh and lateral calf to the dorsum of the foot.
He describes the pain as a deep, burning ache with superimposed electric shock-like sensations that worsen with prolonged standing, walking, and lumbar extension.
On examination, there is weakness of left ankle dorsiflexion and great toe extension (4/5), diminished sensation over the lateral calf and dorsum of the foot, and an absent left Achilles reflex.
Straight leg raise test is positive at 40 degrees on the left, reproducing the radicular symptoms.
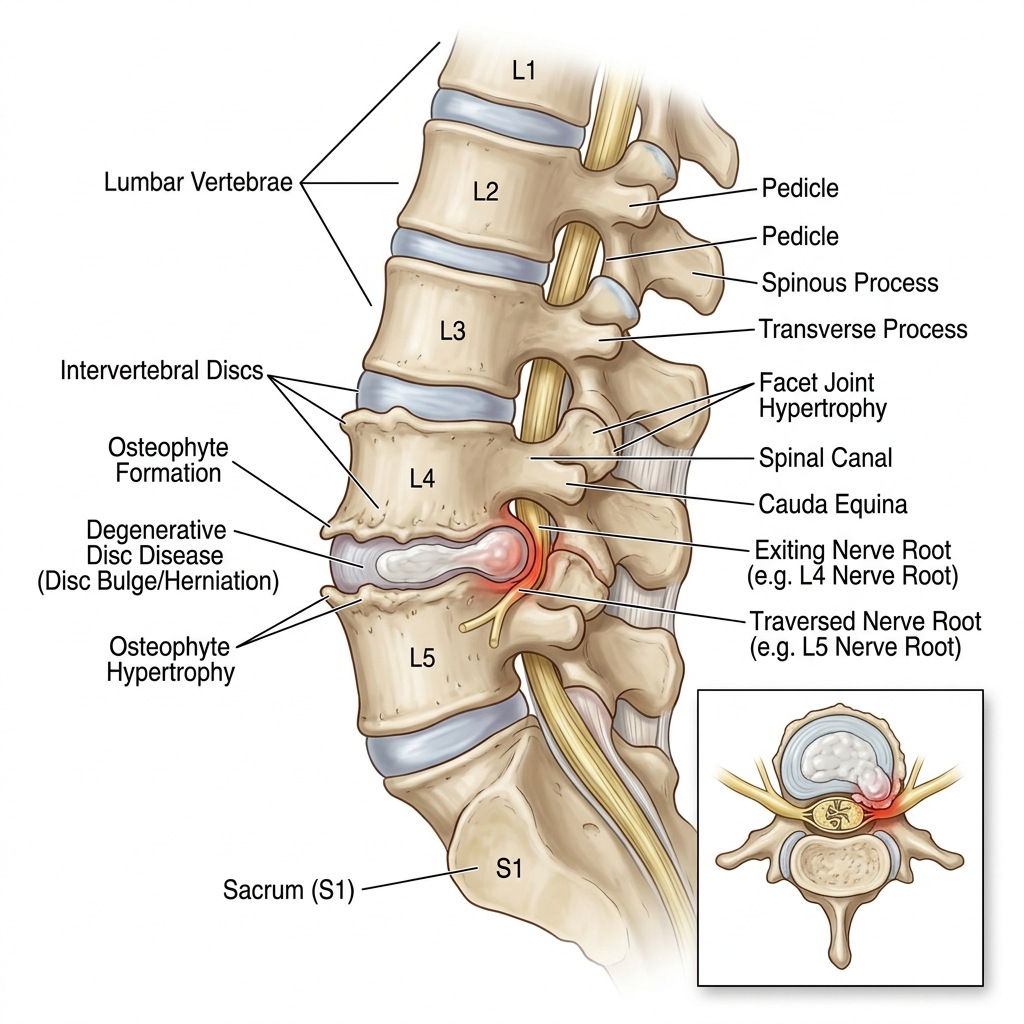
MRI of the lumbar spine demonstrates multilevel degenerative disc disease with a left paracentral L4–L5 disc-osteophyte complex causing L5 nerve root compression and moderate left L5–S1 foraminal stenosis.
Epidemiology
Lumbar spondylosis is a near-universal finding in the aging spine, with radiographic evidence present in more than 90% of individuals over 50 years.
Symptomatic lumbar radiculopathy has a lifetime prevalence of 3–5%, with an annual incidence of approximately 10–25 per 100,000 population.
The L4–L5 and L5–S1 levels account for more than 95% of lumbar radiculopathies, reflecting the biomechanical stress concentrated at the lower lumbar segments.
Risk factors include age, obesity, sedentary lifestyle, heavy manual labor, whole-body vibration exposure, smoking, and prior lumbar spine injury.
Approximately 20–30% of patients with acute lumbar radiculopathy develop chronic symptoms persisting beyond 12 weeks, necessitating ongoing management.
Etiopathophysiology
- Lumbar spondylosis involves progressive degenerative changes affecting the intervertebral discs, facet joints, and ligamentum flavum.
- Disc degeneration leads to loss of disc height, annular bulging, and osteophyte formation at the vertebral endplates and facet joints.
- Foraminal stenosis from a combination of disc bulging, osteophytic overgrowth, facet joint hypertrophy, and ligamentum flavum thickening compresses the exiting nerve roots.
- Central canal stenosis may also contribute, particularly with large midline disc protrusions or diffuse degenerative hypertrophy, causing neurogenic claudication.
- Nerve root compression triggers a cascade of intraneural edema, demyelination, axonal injury, and dorsal root ganglion sensitization.
- Chronic compression activates inflammatory pathways (TNF-α, PGE2, nitric oxide) that sensitize nociceptors and produce neuropathic pain independent of ongoing mechanical compression.
- Central sensitization develops in chronic cases, with amplification of pain signaling at the spinal cord level, contributing to persistent pain and hyperalgesia.
Clinical Features
Patients present with low back pain radiating into the lower extremity in a dermatomal pattern: L4 (anterior thigh, medial leg), L5 (lateral leg, dorsum of foot), or S1 (posterior calf, lateral foot, sole).
The pain is typically described as deep, aching, or burning, often with superimposed sharp, shooting, or electric-like sensations along the affected dermatome.
Motor weakness corresponds to the affected nerve root: L4 (knee extension, hip flexion), L5 (ankle dorsiflexion, great toe extension, hip abduction), S1 (ankle plantarflexion, knee flexion, hip extension).
Sensory loss follows the dermatomal distribution: L4 (medial leg), L5 (lateral leg, web space between 1st and 2nd toes), S1 (lateral foot, sole).
Chronic radiculopathy may manifest as persistent neuropathic pain, muscle atrophy in the affected myotome, reduced functional capacity, and significant psychosocial impact including depression and disability.
Straight leg raise (SLR) test is highly sensitive (~90%) but has modest specificity (~25%) for L4–S1 radiculopathy. Crossed SLR (contralateral leg elevation reproducing ipsilateral symptoms) has much higher specificity (~90%) and strongly suggests disc herniation with nerve root compression. For L3/L4 radiculopathy, the femoral nerve stretch test (prone knee flexion) is the equivalent provocative maneuver.
Diagnosis and Differential Diagnosis
MRI of the lumbar spine is the first-line imaging modality, demonstrating disc degeneration, disc-osteophyte complexes, foraminal or central stenosis, and nerve root compression.
Electrodiagnostic studies (EMG/NCS) are particularly valuable in chronic radiculopathy to confirm the diagnosis, localize the affected root, differentiate from plexopathy or peripheral neuropathy, and assess the degree of axonal loss.
CT myelography is the preferred alternative when MRI is contraindicated and provides excellent delineation of bony foraminal stenosis.
Weight-bearing or dynamic (flexion-extension) radiographs assess alignment, instability, and spondylolisthesis that may contribute to root compression.
Differential diagnoses include lumbosacral plexopathy, piriformis syndrome, hip joint pathology, sacroiliac joint dysfunction, peripheral neuropathy, vascular claudication, and cauda equina syndrome.
Lumbar Root Localization Guide:
| L3 |
L2–L3 |
Anterior thigh |
Hip flexion, knee extension |
Patellar |
Femoral nerve stretch |
| L4 |
L3–L4 |
Anterior thigh, medial leg |
Knee extension, hip adduction |
Patellar |
Femoral nerve stretch |
| L5 |
L4–L5 |
Lateral leg, dorsum of foot |
Ankle dorsiflexion, great toe extension, hip abduction |
None (medial hamstring) |
SLR |
| S1 |
L5–S1 |
Posterior calf, lateral foot, sole |
Ankle plantarflexion, knee flexion |
Achilles |
SLR |
| S2 |
L5–S1 (central) |
Posterior thigh, perineum |
Toe flexors, bladder |
None |
SLR |
- MRI lumbar spine (without contrast initially): First-line to confirm root compression, disc pathology, stenosis; with contrast if tumor or infection suspected.
- EMG/NCS (if >3–4 weeks of symptoms): Localizes affected root, confirms axonal loss, differentiates from plexopathy, peripheral neuropathy, or myopathy.
- Plain radiographs (AP, lateral, flexion-extension): Assess alignment, disc height, spondylolisthesis, instability.
- CT myelography: If MRI contraindicated; excellent for bony detail and lateral recess stenosis.
- Laboratory studies: ESR, CRP if infection/malignancy suspected; HbA1c, B12, TSH if concurrent metabolic neuropathy considered.
Management
Conservative Management (First-line for most cases):
| Physical Therapy |
Core stabilization, lumbar flexion exercises (Williams flexion), nerve mobilization/flossing, directional preference exercises (McKenzie method); 6–12 weeks structured program |
| Pharmacotherapy |
NSAIDs (first-line analgesic), gabapentin/pregabalin for neuropathic pain, short-course oral steroids for acute flares, duloxetine or tricyclics for chronic pain, avoid chronic opioids |
| Lumbar Epidural Steroid Injections |
Transforaminal (preferred for foraminal stenosis) or interlaminar approach; provides 50–70% short-term relief; evidence supports up to 3 injections per year |
| Activity Modification |
Avoid prolonged sitting, proper lifting mechanics, ergonomic workplace assessment, weight management |
| Cognitive Behavioral Therapy |
Recommended for chronic cases; addresses pain catastrophizing, fear avoidance, and functional rehabilitation |
Surgical Management (for refractory or progressive cases):
| Single-level disc herniation |
Microdiscectomy |
Gold standard; >85% success; minimally invasive options available |
| Foraminal stenosis (bony) |
Laminectomy/foraminotomy ± fusion |
Decompression with or without instrumented fusion |
| Multilevel stenosis with instability |
Laminectomy + posterior lumbar interbody fusion (PLIF) or transforaminal lumbar interbody fusion (TLIF) |
For stenosis with spondylolisthesis or instability |
| Recurrent disc herniation |
Revision microdiscectomy ± fusion |
Fusion considered if >2 recurrences or significant instability |
| Failed conservative therapy >6–12 weeks |
Surgical consultation |
Progressive motor deficit, cauda equina syndrome, or intractable pain |
Management Algorithm: 1. Initial presentation: Confirm diagnosis with MRI ± EMG; initiate conservative therapy (NSAIDs, structured PT, activity modification). 2. 4–6 weeks: Reassess; if inadequate response, add neuropathic pain agents (gabapentin/pregabalin), consider lumbar epidural steroid injection. 3. 6–12 weeks: If refractory, repeat imaging, advanced electrodiagnostics; surgical consultation for progressive weakness, cauda equina symptoms, or significant functional impairment. 4. Surgical candidates: Microdiscectomy for contained disc herniation; decompression ± fusion for stenosis/instability; postoperative rehabilitation for 6–12 weeks. 5. Long-term: Core strengthening, weight management, ergonomic optimization, periodic reassessment for adjacent segment degeneration or recurrent stenosis.
Multiple Choice Question
A 60-year-old woman presents with 5 months of left-sided low back pain radiating to the posterior calf and lateral foot, with weakness of ankle plantarflexion and an absent left Achilles reflex. MRI shows L5–S1 foraminal stenosis. Which nerve root is most likely compressed?
- L3
- L4
- L5
- S1
- S2
Answer: (D) S1 radiculopathy presents with pain radiating to the posterior calf, lateral foot, and sole; weakness of ankle plantarflexion (and knee flexion); and diminished or absent Achilles reflex. The L5–S1 disc level compresses the exiting S1 nerve root.
References
- Kreiner DS, Hwang SW, Easa JE, et al. An evidence-based clinical guideline for the diagnosis and treatment of lumbar disc herniation with radiculopathy. Spine J. 2014;14(1):180–191. DOI: 10.1016/j.spinee.2013.08.003
- Tarulli AW, Raynor EM. Lumbosacral radiculopathy. Neurol Clin. 2007;25(2):387–405. DOI: 10.1016/j.ncl.2007.01.008
- Patel EA, Perloff MD. Radicular pain syndromes: cervical, lumbar, and spinal stenosis. Semin Neurol. 2018;38(6):634–639. DOI: 10.1055/s-0038-1673680
- Ropper AH, Zafonte RD. Sciatica. N Engl J Med. 2015;372(13):1240–1248. DOI: 10.1056/NEJMra1410151
- Chou R, Hashimoto R, Friedly J, et al. Epidural corticosteroid injections for radiculopathy and spinal stenosis: a systematic review and meta-analysis. Ann Intern Med. 2015;163(5):373–381. DOI: 10.7326/M15-0934
Migraine

📊 Quick Facts
- Prevalence: 12–15% worldwide
- Gender: Female:Male ≈ 3:1 (childhood ratio equalizes)
- Peak Age: 25–45 years
- Aura Frequency: 25–30% of patients
- Chronic Migraine: ≥15 headache days/month (~2% of population)
⚠️ Red Flags
- Thunderclap onset → Evaluate for SAH/cerebral venous thrombosis
- New headache >50 yrs → Image to exclude secondary causes
- Focal deficits or seizures atypical for aura → Urgent imaging
- Systemic symptoms (fever, weight loss) → Consider infection/vasculitis
- Neck stiffness, papilledema → Rule out meningitis/ICP elevation
Clinical Scenario
A 32-year-old woman presents with recurrent, throbbing headaches over the past 8 years, typically unilateral and associated with photophobia, phonophobia, and nausea.
Attacks last between 4 and 72 hours and are often preceded by visual scintillations and zigzag lines lasting about 20 minutes.
The headaches significantly interfere with her daily functioning and are aggravated by physical activity.
There is a strong family history of similar headaches in her mother and sister.
These findings are consistent with migraine with aura, a primary headache disorder characterized by episodic neurovascular dysfunction.
Epidemiology
Migraine is a common primary headache disorder affecting approximately 12–15% of the global population.
It is more prevalent in women (3:1 female-to-male ratio) and typically begins in adolescence or early adulthood.
The peak incidence occurs between 25 and 45 years of age, coinciding with the most productive years of life.
Genetic predisposition plays a major role, with up to 70% of patients having a positive family history.
Migraine is a leading cause of disability worldwide and contributes substantially to socioeconomic burden due to lost productivity.
Etiopathophysiology
Cortical spreading depression generates aura and activates brainstem nuclei (dorsal pons), which in turn excite the trigeminovascular system.
Trigeminal afferents release CGRP, substance P, and neurokinin A, producing vasodilation, plasma extravasation, and neurogenic inflammation.
Central sensitization within the trigeminal nucleus caudalis and thalamus explains cutaneous allodynia and photophobia.
Hormonal fluctuations, genetic ion-channel variants (CACNA1A, SCN1A), and environmental triggers modulate neuronal excitability.
CGRP monoclonal antibodies and gepants directly target this pathway, validating the mechanistic model.
Clinical Features
Migraine typically presents as recurrent, unilateral, pulsating headaches lasting 4–72 hours, often accompanied by nausea, vomiting, photophobia, and phonophobia.
Aura occurs in about 25–30% of patients and may manifest as visual disturbances (scintillating scotomas, fortification spectra), sensory symptoms, or dysphasia.
The attacks are often preceded by premonitory symptoms (e.g., yawning, mood changes) and followed by a postdromal phase with fatigue or cognitive slowing.
Physical activity, hormonal changes, stress, certain foods, and sleep disturbances are common triggers.
Neurological examination is usually normal between attacks, and chronic migraine is defined as headaches occurring on 15 or more days per month for over 3 months.
Positive, spreading aura lasting 5–60 minutes followed by headache strongly favors migraine. Sudden negative symptoms (vision loss, weakness) without headache should prompt vascular evaluation.
Diagnosis and Differential Diagnosis
Diagnosis is clinical and based on the International Classification of Headache Disorders (ICHD-3) criteria.
Typical features include recurrent unilateral pulsating headaches with moderate to severe intensity, aggravated by routine activity, and associated with nausea or sensory sensitivities.
Neuroimaging is indicated in atypical presentations, new-onset headache after age 50, focal neurological deficits, or red flag symptoms (e.g., papilledema, seizures).
Differential diagnoses include tension-type headache, cluster headache, trigeminal neuralgia, idiopathic intracranial hypertension, and secondary causes such as intracranial mass or vascular malformation.
Distinguishing migraine aura from transient ischemic attack (TIA) is critical; aura develops gradually and is often positive (e.g., visual scintillations), whereas TIA is sudden and negative (e.g., vision loss).
Differential Diagnosis Comparison:
| Pain Quality |
Pulsating, moderate-severe |
Pressing, mild-moderate |
Excruciating orbital |
Pressure/daily diffuse |
| Duration |
4–72 h |
30 min–7 d |
15–180 min |
Daily, persistent |
| Laterality |
Unilateral (can be bilateral) |
Bilateral |
Strictly unilateral |
Diffuse |
| Associated Symptoms |
Nausea, photophobia, aura |
None/minimal |
Autonomic (tearing, ptosis) |
Papilledema, visual obscurations |
| Triggers |
Hormonal, foods, stress |
Stress, posture |
Alcohol, nocturnal onset |
Obesity, medications |
Migraine without aura: ≥5 attacks lasting 4–72 h with ≥2 of: unilateral, pulsating, moderate/severe, aggravated by activity and ≥1 of nausea/vomiting or photophobia/phonophobia.
Migraine with aura: ≥2 attacks with reversible aura symptoms (visual, sensory, speech, motor, brainstem, retinal) lasting 5–60 min, followed by headache within 60 min.
Exclude other diagnoses (SNOOP red flags) via history/exam ± imaging.
Management
Acute Therapy (treat within 1 hour of onset):
| NSAIDs / Acetaminophen |
Ibuprofen 400-800 mg, naproxen 500-550 mg |
First-line for mild-moderate attacks |
| Triptans |
Sumatriptan 50-100 mg PO or 6 mg SC; rizatriptan 10 mg |
Avoid in CAD, uncontrolled HTN; combine with NSAID for synergy |
| Gepants |
Ubrogepant 50-100 mg, rimegepant 75 mg ODT |
Useful when triptans contraindicated |
| Ditans |
Lasmiditan 50-200 mg |
Non-vasoconstrictive; caution driving for 8 h |
| Adjuncts |
Metoclopramide, prochlorperazine |
Address nausea, enhance absorption |
Preventive Therapy (≥4 migraine days/month or disabling attacks):
| Beta-blockers |
Propranolol, metoprolol |
Grade A |
Avoid in asthma, bradycardia |
| Antiepileptics |
Topiramate, valproate |
Grade A |
Topiramate causes paresthesia/weight loss; valproate teratogenic |
| Antidepressants |
Amitriptyline, venlafaxine |
Grade B |
Helpful with comorbid insomnia, depression |
| CGRP mAbs |
Erenumab, fremanezumab, galcanezumab, eptinezumab |
Grade A |
Monthly/quarterly injections; minimal systemic effects |
| OnabotulinumtoxinA |
155-195 U q12wk |
Grade A for chronic migraine |
PREEMPT injection protocol; safe with CGRP mAbs |
| Neuromodulation |
Single-pulse TMS, vagal nerve stimulation |
Grade C |
Adjunct for refractory patients |
Treatment Algorithm: 1. Assess frequency & disability (MIDAS, HIT-6). 2. Acute plan: Educate on early dosing, limit acute meds to ≤10 days/month to avoid medication-overuse headache. 3. Preventive plan: Start first-line agent; titrate every 4-6 weeks; set goals (≥50% reduction in migraine days). 4. Refractory cases: Combine preventive classes (e.g., CGRP mAb + onabotulinumtoxinA), refer to headache specialist, consider lifestyle coaching, CBT, or biofeedback. 5. Lifestyle: Regular sleep, hydration, exercise, trigger diary, mindful stress reduction.
Multiple Choice Question
Which of the following features most strongly supports a diagnosis of migraine with aura rather than transient ischemic attack (TIA)?
- Sudden onset of visual field loss without positive phenomena
- Gradual progression of visual scintillations followed by headache
- Unilateral headache lasting less than 30 minutes
- Complete resolution of symptoms within 5 minutes
Answer: (B) Gradual progression of visual scintillations followed by headache Migraine aura typically develops gradually over 5–20 minutes,is often positive (e.g., visual scintillations), and is followed by a headache, whereas TIA is abrupt, negative, and often not followed by headache.
References
Goadsby PJ, Holland PR, Martins-Oliveira M, Hoffmann J, Schankin C, Akerman S. Pathophysiology of migraine: a disorder of sensory processing. Physiol Rev. 2017;97(2):553–622. DOI: 10.1152/physrev.00034.2015
Charles A. The pathophysiology of migraine: implications for clinical management. Lancet Neurol. 2018;17(2):174–182. DOI: 10.1016/S1474-4422(17)30435-0
Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition.Cephalalgia. 2018;38(1):1–211. DOI: 10.1177/0333102417738202
Myasthenia Gravis

📊 Quick Facts
- Prevalence: 20-50 per 100,000
- Peak Age: Bimodal (F: 20-40, M: 60-80 years)
- Antibodies: AChR (80-85%), MuSK (5-8%)
- Thymic Abnormalities: 75% (hyperplasia 65%, thymoma 10-15%)
- Myasthenic Crisis: 15-20% lifetime risk
⚠️ Red Flags
- Respiratory distress → Myasthenic crisis (ICU needed)
- Negative AChR + MuSK → Consider seronegative MG or alternative diagnosis
- Thymoma on CT → Urgent thymectomy consultation
- Worsening with cholinesterase inhibitors → Cholinergic crisis
- Contraindicated drugs → Aminoglycosides, beta-blockers, fluoroquinolones
Clinical Scenario
A 32-year-old woman presents with progressive double vision and drooping eyelids that worsen by evening and improve with rest.
Over several weeks, she develops difficulty chewing and swallowing, with occasional shortness of breath.
Neurological examination reveals fatigable weakness of the extraocular muscles and proximal limb muscles, with preserved reflexes and sensation.
Administration of edrophonium (a short-acting acetylcholinesterase inhibitor) produces transient improvement in muscle strength.
These findings are consistent with myasthenia gravis (MG), an autoimmune disorder affecting the neuromuscular junction.
Epidemiology
MG has an estimated prevalence of 20–50 per 100,000 population worldwide, with incidence rates of 3–5 per million per year.
The disease shows a bimodal age distribution: young women (20–40) years) and older men (60–80 years).
There is no significant ethnic predilection, although incidence may vary geographically.
Thymic abnormalities, including thymic hyperplasia (65%) and thymoma (10–15%), are common associations.
Advances in diagnosis and treatment have significantly improved survival, with most patients achieving near-normal life expectancy.
Etiopathophysiology
MG is an autoimmune disease characterized by antibodies targeting components of the postsynaptic neuromuscular junction.
Most commonly, antibodies are directed against the acetylcholine receptor (AChR); others include muscle-specific kinase (MuSK) and low-density lipoprotein receptor-related protein 4 (LRP4).
These autoantibodies reduce functional AChR density through complement-mediated destruction, receptor internalization, and blockade of receptor function.
The result is impaired neuromuscular transmission, manifesting as fluctuating muscle weakness and fatigability.
Thymic abnormalities contribute to autoimmune activation by supporting autoreactive T-cell development.
Key Mechanisms: - AChR antibody (80-85%): Complement-mediated receptor destruction - MuSK antibody (5-8%): Impaired receptor clustering, IgG4-mediated (no complement) - LRP4 antibody (<5%): Disrupts agrin-LRP4-MuSK signaling - Thymic role: Hyperplasia or thymoma drives autoimmune response - Decremental response: Reduced ACh quanta → fatigue with repetition
Clinical Features
The hallmark feature is fluctuating muscle weakness that worsens with exertion and improves with rest (“fatigability”).
Ocular involvement occurs in over 50% of patients, presenting with ptosis and diplopia. 15% remain purely ocular.
Bulbar symptoms such as dysarthria, dysphagia, and chewing fatigue are common in generalized MG.
Limb and axial muscle weakness can occur, typically affecting proximal muscles more than distal ones.
Respiratory muscle involvement can lead to life-threatening myasthenic crisis, often precipitated by infection or medication.
Classic triad: Ptosis + diplopia + fatigable weakness that worsens by evening and improves with rest. Ice pack test for ptosis (>2mm improvement) is 80% sensitive. Edrophonium test is obsolete (cardiac risks). MuSK-positive MG → predominantly bulbar/facial weakness, less ocular involvement, poorer response to cholinesterase inhibitors.
Diagnosis and Differential Diagnosis
Diagnostic Approach: - Diagnosis is based on clinical features, antibody testing, neurophysiological studies, and pharmacologic response.
- Serum antibodies:
- AChR antibodies: 80-85% of generalized MG, 50% of ocular MG
- MuSK antibodies: 5-8% (AChR-negative generalized MG)
- LRP4 antibodies: <5% (double seronegative MG)
- Seronegative MG: 10-15% (all antibodies negative)
- Neurophysiological studies:
- Repetitive nerve stimulation (RNS): >10% decrement at 3 Hz (sensitivity 50-75%)
- Single-fiber EMG: Increased jitter and blocking (sensitivity 95-99%, gold standard)
- Clinical tests:
- Ice pack test: >2 mm ptosis improvement (sensitivity 80%, for ocular MG)
- Edrophonium test: Obsolete (cardiac risks)
Differential Diagnosis Comparison:
| Weakness Pattern |
Ocular > bulbar > limb |
Proximal limb > ocular rare |
Descending (cranial → limb) |
Asymmetric, progressive |
| Fatigability |
Worsens with use |
Improves with use |
Stable/worsens |
Progressive, no fatigue |
| Autonomic |
Absent |
Dry mouth, impotence |
Dilated pupils, ileus |
Absent |
| Reflexes |
Normal/brisk |
Reduced, post-exercise facilitation |
Reduced/absent |
Hyperreflexia + Babinski |
| EMG Pattern |
Decremental (RNS) |
Incremental >100% (RNS) |
Decremental |
Fibrillations, fasciculations |
| Antibodies |
AChR (80-85%), MuSK (5-8%) |
P/Q-type VGCC (85-90%) |
None (toxin assay) |
None |
| Association |
Thymus (75%) |
Small cell lung cancer (60%) |
Contaminated food |
None |
Definite MG requires ONE of: 1. Positive antibodies: AChR, MuSK, or LRP4 2. Abnormal neurophysiology + clinical improvement with cholinesterase inhibitors: - RNS with >10% decrement, OR - Single-fiber EMG with increased jitter/blocking 3. Clinical improvement with immunotherapy in seronegative cases
Supporting features: - Fluctuating fatigable weakness - Ocular and/or bulbar involvement - Improvement with rest - Thymic abnormality on CT chest
Management
Symptomatic Treatment (Cholinesterase Inhibitors):
| Pyridostigmine |
30-120 mg q4-6h (max 600 mg/day) |
PO |
30-60 min |
Grade A |
First-line symptomatic; avoid overdose (cholinergic crisis) |
| Neostigmine |
15 mg q4h |
PO |
45-75 min |
Grade B |
Alternative to pyridostigmine |
Immunosuppressive Therapy (Long-term Control):
| Prednisone |
0.5-1 mg/kg/day (start 10-20 mg, titrate up) |
2-4 weeks |
Grade A |
Taper after improvement; initial worsening possible |
| Azathioprine |
2-3 mg/kg/day (max 200 mg) |
6-12 months |
Grade A |
Steroid-sparing; monitor LFTs, CBC |
| Mycophenolate |
1,000-1,500 mg BID |
6-12 months |
Grade B |
Better tolerated than azathioprine; teratogenic |
| Cyclosporine |
3-5 mg/kg/day divided BID |
4-8 weeks |
Grade B |
Faster onset; monitor BP, creatinine |
| Tacrolimus |
3-5 mg/day |
4-8 weeks |
Grade C |
Alternative to cyclosporine |
| Rituximab |
375 mg/m² weekly × 4, or 1 g × 2 |
3-6 months |
Grade B |
Especially MuSK+ MG; monitor infections |
Rapid Immunomodulation (Crisis/Severe Exacerbation):
| IVIG |
2 g/kg over 2-5 days |
Grade A |
Myasthenic crisis, severe exacerbation, preoperative |
| Plasma Exchange (PLEX) |
5 exchanges over 10-14 days |
Grade A |
Myasthenic crisis, refractory cases |
| Eculizumab |
900 mg weekly × 4, then 1,200 mg q2 weeks |
Grade B |
Refractory generalized AChR+ MG (complement inhibitor) |
Surgical Treatment:
| Thymoma |
Urgent (malignancy risk) |
Grade A |
Tumor control; variable MG improvement |
| Generalized AChR+ MG (age <60) |
Within 2 years of onset |
Grade A |
50-60% remission/minimal manifestation at 3 years |
| Pure ocular MG |
Not indicated |
Grade C |
Low benefit |
Treatment Algorithm:
- Ocular MG (Purely):
- Pyridostigmine 30-60 mg q6h [Grade A]
- If inadequate → add prednisone (start 10-20 mg/day) [Grade B]
- Consider thymectomy if age <60 and AChR+ [Grade C, Weak]
- Mild Generalized MG:
- Pyridostigmine up to 120 mg q4h [Grade A]
- CT chest to evaluate thymus
- Consider thymectomy if age <60 and AChR+ [Grade A]
- If inadequate control → add prednisone 0.5-1 mg/kg/day [Grade A]
- Moderate-Severe Generalized MG:
- Prednisone 0.5-1 mg/kg/day + pyridostigmine [Grade A]
- Add steroid-sparing agent early (azathioprine or mycophenolate) [Grade A]
- Thymectomy if age <60 and AChR+ [Grade A]
- If refractory → rituximab (especially MuSK+) [Grade B]
- Myasthenic Crisis (Respiratory Failure):
- ICU admission with intubation if FVC <15 mL/kg or NIF >-30 cmH₂O
- IVIG (2 g/kg) OR PLEX (5 exchanges) [Grade A]
- Hold cholinesterase inhibitors (avoid cholinergic crisis)
- Start high-dose steroids after IVIG/PLEX [Grade A]
- Treat precipitant (infection, medication)
- Refractory MG (Failed ≥2 immunosuppressants):
- Rituximab [Grade B]
- Eculizumab (if AChR+ and complement-mediated) [Grade B]
- Chronic IVIG or PLEX maintenance
Medications to AVOID in MG: - ❌ Antibiotics: Aminoglycosides, fluoroquinolones, macrolides - ❌ Cardiac: Beta-blockers, calcium channel blockers, quinidine - ❌ Neuromuscular: Muscle relaxants, magnesium - ❌ Other: D-penicillamine, interferon-alpha, checkpoint inhibitors
Multiple Choice Question
Which of the following best distinguishes myasthenia gravis from Lambert-Eaton myasthenic syndrome (LEMS)?
- Presence of proximal muscle weakness
- Presence of ocular symptoms such as ptosis and diplopia
- Improvement of strength with repeated stimulation
- Association with malignancy
Answer: (B) Presence of ocular symptoms such as ptosis and diplopia Ocular involvement is characteristic of MG and rare in LEMS, which typically improves with repeated activity due to presynaptic facilitation.
References
- Gilhus NE, Tzartos S, Evoli A, Palace J, Burns TM, Verschuuren JJ. Myasthenia gravis. Nat Rev Dis Primers. 2019;5(1):30. DOI: 10.1038/s41572-019-0079-y
- Drachman DB. Myasthenia gravis. N Engl J Med. 1994;330(25):1797–1810. DOI: 10.1056/NEJM199406233302507
- Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for management of myasthenia gravis. Neurology. 2016;87(4):419–425. DOI: 10.1212/WNL.0000000000002790
Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease (MOGAD)
Clinical Scenario
A 28-year-old woman presents with sudden onset bilateral vision loss over three days, preceded by eye pain on movement.
MRI of the brain and orbits shows bilateral optic nerve enhancement without significant demyelinating lesions elsewhere.
Serum testing is positive for myelin oligodendrocyte glycoprotein (MOG)-IgG antibodies using a live cell-based assay.
Over the next few weeks, she develops new neurological deficits including lower limb weakness and bladder dysfunction.
These findings are consistent with MOGAD, an inflammatory demyelinating disease distinct from multiple sclerosis (MS) and neuromyelitis optica spectrum disorder (NMOSD).
Epidemiology
MOGAD is a rare autoimmune demyelinating disease of the central nervous system (CNS), accounting for ~5–10% of cases initially suspected to be MS or NMOSD.
It affects both children and adults, with a slight male predominance in pediatric cases and a female predominance in adults.
The mean age of onset is between 20 and 40 years, though cases can occur across the lifespan.
Pediatric cases often present as acute disseminated encephalomyelitis (ADEM), while adults frequently present with optic neuritis or transverse myelitis.
Relapsing disease occurs in about 30–50% of patients, but overall prognosis is better than MS or AQP4-positive NMOSD.
Etiopathophysiology
MOGAD is mediated by pathogenic IgG1 antibodies directed against myelin oligodendrocyte glycoprotein, a surface protein on CNS myelin.
These antibodies trigger complement activation, antibody-dependent cellular cytotoxicity, and demyelination.
Unlike MS, MOGAD lacks significant T-cell infiltration and chronic neurodegeneration.
It is considered a distinct nosological entity from AQP4-positive NMOSD, which targets astrocytic aquaporin-4 channels.
The disease is monophasic in many patients, though recurrent episodes are possible, especially without long-term immunotherapy.
Clinical Features
Common presentations include optic neuritis (often bilateral), transverse myelitis (usually longitudinally extensive), and ADEM-like encephalitis (especially in children).
Optic neuritis in MOGAD typically shows marked optic disc swelling and severe visual loss, but visual recovery is often good.
Myelitis may cause acute paraparesis or quadriparesis, sensory level, and autonomic dysfunction.
Brain involvement can mimic MS but often has ill-defined or fluffy lesions, sometimes involving the deep gray matter.
Relapses, if they occur, are often milder and more steroid-responsive compared to AQP4-NMOSD.
Diagnosis and Differential Diagnosis
Diagnosis is based on a compatible clinical syndrome, MRI findings, and detection of serum MOG-IgG using a live cell-based assay.
CSF may show mild pleocytosis and elevated protein but rarely has oligoclonal bands.
MRI findings include longitudinally extensive spinal cord lesions and optic nerve enhancement often involving the anterior segments.
Differentials include multiple sclerosis, AQP4-NMOSD, infectious myelitis, and paraneoplastic demyelination.
AQP4-IgG and MOG-IgG should both be tested to accurately differentiate MOGAD from NMOSD.
Management
Acute treatment involves high-dose intravenous corticosteroids,followed by a slow taper over weeks.
Plasma exchange or IVIG is used for severe or steroid-refractory attacks.
Maintenance therapy is considered in relapsing disease and may includem azathioprine, mycophenolate mofetil, rituximab, or IVIG.
Long-term immunotherapy is often not required for monophasic cases.
Regular monitoring with MRI and antibody titers can help guide treatment duration and relapse prevention.
Multiple Choice Question
Which of the following features most reliably distinguishes MOGAD from AQP4-positive NMOSD?
- Presence of longitudinally extensive transverse myelitis
- Severe optic neuritis
- Good visual recovery and steroid responsiveness
- Female predominance
Answer: (C) Good visual recovery and steroid responsiveness While both diseases can cause optic neuritis and extensive myelitis, MOGAD is characterized by better recovery and higher responsiveness to corticosteroids compared to AQP4-positive NMOS
References
- Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol. 2019;15(2):89–102. DOI: 10.1038/s41582-018-0112-x
- Cobo-Calvo Á, Ruiz A, Maillart E, et al. Clinical spectrum and prognostic value of MOG-antibody–associated disease in adults. Neurology. 2018;90(21):e1858–e1869. DOI: 10.1212/WNL.0000000000005560
- Sato DK, Callegaro D, Lana-Peixoto MA, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. JAMA Neurol. 2014;71(3):276–283. DOI: 10.1001/jamaneurol.2013.5857
Paraneoplastic Neurologic Syndromes (PNS)

- Incidence: <1% of cancer patients; often precedes cancer diagnosis
- Most common cancers: SCLC, breast, ovarian, thymic, testicular
- Antibody detection: 60-70% cases; guides cancer search
- Classic syndromes: Limbic encephalitis, cerebellar degeneration, sensory neuronopathy
- Key distinction: Intracellular Ab (poor prognosis) vs. surface Ab (better response)
- Treatment: Treat underlying cancer + immunotherapy
- Subacute neurologic decline in patient with cancer history or risk factors
- Unexplained ataxia, seizures, or memory loss - consider occult malignancy
- Anti-Hu antibodies - high association with SCLC (search aggressively)
- Rapidly progressive symptoms despite treatment - suggests T-cell mediated
- Multifocal neurologic involvement - PNS can affect multiple nervous system levels
- New psychiatric symptoms in older adults - may be limbic encephalitis
- Positive onconeural antibodies - mandate tumor search even if imaging negative
Clinical Scenario
A 64-year-old woman with 40-pack-year smoking history presents with progressive gait ataxia, diplopia, and new-onset short-term memory impairment over 3 months. Neurological examination reveals limb dysmetria, truncal instability, and mild confusion, but no focal motor or sensory deficits. Brain MRI is unremarkable, and CSF shows mild lymphocytic pleocytosis without evidence of infection or malignancy. Serum testing reveals anti-Hu (ANNA-1) antibodies. Subsequent CT chest identifies small-cell lung carcinoma, leading to diagnosis of paraneoplastic limbic encephalitis and cerebellar degeneration.
Epidemiology
- Rare immune-mediated disorders; occur in <1% of cancer patients
- Most common associated cancers: SCLC (most common), breast, ovarian, thymic, testicular germ cell tumors
- Can affect any nervous system level: CNS, PNS, autonomic, neuromuscular junction
- Onconeural antibodies detected in 60-70% cases; often precede cancer diagnosis by months to years
- Early recognition critical - neurologic symptoms may be presenting feature of occult malignancy
Immune-mediated mechanism: - Immune response against shared antigens (onconeural antigens) expressed by tumor and neural tissue - Tumor-induced B and T lymphocyte activation → autoantibody production + cytotoxic neuronal damage - Cross-reactivity between tumor antigens and neuronal proteins
Antibody classification and prognosis:
1. Intracellular (cytoplasmic/nuclear) antibodies: - Examples: Anti-Hu (ANNA-1), Anti-Yo (PCA-1), Anti-Ri (ANNA-2), Anti-Ma2 - Mechanism: T-cell mediated neuronal destruction - Prognosis: Poor; irreversible neuronal damage - Treatment response: Limited response to immunotherapy - Associated syndromes: Limbic encephalitis, cerebellar degeneration, sensory neuronopathy
2. Cell-surface/synaptic antibodies: - Examples: Anti-NMDA receptor, Anti-LGI1, Anti-CASPR2, Anti-AMPA receptor - Mechanism: Antibody-mediated reversible synaptic dysfunction - Prognosis: Better; potential for recovery - Treatment response: More likely to respond to immunotherapy - Associated syndromes: Encephalitis, seizures, movement disorders
Common antibody-cancer-syndrome associations: - Anti-Hu (ANNA-1): SCLC → limbic encephalitis, sensory neuronopathy, encephalomyelitis - Anti-Yo (PCA-1): Ovarian/breast cancer → cerebellar degeneration - Anti-Ri (ANNA-2): Breast/SCLC → brainstem encephalitis, cerebellar ataxia - Anti-Ma2: Testicular germ cell tumor → limbic/brainstem encephalitis - Anti-CV2 (CRMP5): SCLC/thymoma → encephalomyelitis, chorea, uveitis
Clinical Features
General characteristics: - Symptoms may precede tumor diagnosis by months to years - Typically subacute onset and progressive course (distinguishes from metabolic/infectious causes) - Can affect any level of nervous system
Central nervous system syndromes:
Limbic encephalitis: - Short-term memory loss, confusion - Seizures (temporal lobe) - Psychiatric changes (personality, mood, hallucinations) - MRI: T2/FLAIR hyperintensity in medial temporal lobes
Cerebellar degeneration: - Progressive ataxia (gait, limb, truncal) - Dysarthria - Nystagmus - MRI: cerebellar atrophy (late finding)
Brainstem encephalitis: - Cranial neuropathies - Nystagmus, vertigo - Dysphagia, dysarthria - Respiratory compromise (severe cases)
Encephalomyelitis: - Multifocal CNS involvement - Cognitive, motor, sensory deficits - Seizures
Peripheral nervous system syndromes:
Sensory neuronopathy (subacute sensory ganglionopathy): - Asymmetric sensory loss (pain, proprioception) - Sensory ataxia - Preserved motor function - Associated with anti-Hu
Motor neuropathy: - Guillain-Barré-like presentation - Progressive weakness - Areflexia
Autonomic dysfunction: - Orthostatic hypotension - Gastrointestinal dysmotility - Bladder dysfunction - Cardiac arrhythmias
Neuromuscular junction: - Lambert-Eaton myasthenic syndrome (LEMS) - SCLC - Myasthenia gravis - thymoma
Diagnosis
Diagnostic criteria (2021 updated criteria): Requires clinical syndrome + onconeural antibody and/or cancer within 5 years
Clinical evaluation: - Detailed neurological examination - Subacute progressive symptoms (<3 months to peak) - Exclude other causes
Antibody testing: - Serum and CSF onconeural antibodies - High-risk antibodies: Anti-Hu, Anti-Yo, Anti-Ri, Anti-Ma2, Anti-CV2 - Intermediate-risk: Anti-SOX1, Anti-Tr, Anti-Zic4 - Send comprehensive paraneoplastic panel
Neuroimaging: - MRI brain/spine with contrast - Limbic encephalitis: T2/FLAIR hyperintensity in medial temporal lobes - Cerebellar degeneration: atrophy (late finding) - Often normal or subtle findings - FDG-PET brain: increased metabolism in affected regions
CSF analysis: - Lymphocytic pleocytosis (10-100 cells/μL) - Elevated protein - Oligoclonal bands - Normal glucose - Negative infectious workup
Cancer screening: - Comprehensive tumor search mandatory: - CT chest/abdomen/pelvis with contrast - FDG-PET/CT (most sensitive for occult malignancy) - Mammography, pelvic ultrasound (women) - Testicular ultrasound (men) - Repeat screening every 3-6 months if initial negative (up to 4 years) - Antibody type guides cancer search: - Anti-Hu → SCLC (chest CT) - Anti-Yo → ovarian/breast cancer - Anti-Ma2 → testicular germ cell tumor
Differential diagnosis: - Primary autoimmune encephalitis (without cancer) - Metastatic disease to CNS - Parainfectious/post-infectious encephalitis - Toxic/metabolic encephalopathy - Neurodegenerative disease (Alzheimer’s, CJD) - CNS lymphoma - Wernicke encephalopathy
Management
Principle: Treat cancer + immunotherapy. Prognosis depends on antibody type and cancer treatment success.
Cancer treatment (cornerstone): - Identify and treat underlying malignancy promptly - Tumor-directed therapy often stabilizes or improves neurologic symptoms - Surgery, chemotherapy, radiation as appropriate for cancer type - Even partial tumor response may benefit neurologic symptoms
Immunotherapy:
First-line (especially for surface antibody syndromes): - Corticosteroids: methylprednisolone 1 g IV daily × 3-5 days, then oral taper - IVIG: 2 g/kg divided over 2-5 days - Plasma exchange (PLEX): 5-7 exchanges over 10-14 days - Often use combination approach
Second-line (refractory cases): - Rituximab: 375 mg/m² weekly × 4 doses (anti-CD20) - Cyclophosphamide: 750 mg/m² monthly - Mycophenolate mofetil: 1000-1500 mg twice daily - Tocilizumab: for refractory anti-NMDA receptor encephalitis
Symptomatic management: - Seizures: antiepileptic drugs (levetiracetam, lacosamide) - Ataxia: physical therapy, assistive devices - Dysautonomia: fludrocortisone, midodrine, salt supplementation - Cognitive/psychiatric: supportive care, cognitive rehabilitation - Pain: neuropathic pain management (gabapentin, pregabalin)
Monitoring: - Serial neurologic examinations - Repeat MRI to assess response - Monitor for cancer recurrence - Long-term follow-up essential
Prognosis:
Surface antibody syndromes: - Better prognosis - Potential for significant recovery with treatment - Examples: Anti-NMDA receptor, Anti-LGI1
Intracellular antibody syndromes: - Poor prognosis - Irreversible neuronal damage (T-cell mediated) - Incomplete recovery despite treatment - Examples: Anti-Hu, Anti-Yo, Anti-Ri
Factors predicting outcome: - Early cancer detection and treatment - Antibody type (surface vs. intracellular) - Severity at presentation - Promptness of immunotherapy initiation
Multiple Choice Question
oindent Which of the following antibodies is most commonly associated with paraneoplastic cerebellar degeneration in a patient with breast cancer?
- Anti-Hu (ANNA-1)
- Anti-Yo (PCA-1)
- Anti-Ri (ANNA-2)
- Anti-Ma2
Answer: (B) Anti-Yo (PCA-1)
References
- Graus F, Vogrig A, Muñiz-Castrillo S, et al. Updated diagnostic criteria for paraneoplastic neurologic syndromes. Neurol Neuroimmunol Neuroinflamm. 2021;8(4):e1014. DOI: 10.1212/NXI.0000000000001014.
- Darnell RB, Posner JB. Paraneoplastic syndromes affecting the nervous system. Semin Oncol. 2006;33(3):270–298. DOI: 10.1053/j.seminoncol.2006.03.008.
- Lancaster E. The paraneoplastic disorders. Continuum (Minneap Minn). 2015;21(2 Neuro-oncology):452–475. DOI: 10.1212/01.CON.0000464180.89580.88.
Parkinson’s Disease

📊 Quick Facts
- Prevalence: 100-200 per 100,000 (1-2%)
- Peak Age: ~60 years (onset >50)
- Gender: M>F (slight predominance)
- Course: Progressive, chronic
- Genetics: 10-15% familial (LRRK2, SNCA, PARK7)
⚠️ Red Flags
- Symmetric onset → Consider atypical parkinsonism (MSA, PSP)
- Poor levodopa response → Not idiopathic PD (likely PSP/MSA)
- Early falls (within 1 year) → Progressive supranuclear palsy
- Early autonomic failure → Multiple system atrophy
- Rapid cognitive decline → Dementia with Lewy bodies
Clinical Scenario
A 68-year-old man presents with a 2-year history of progressive slowness of movement and stiffness, initially affecting his right hand.
His wife reports a resting tremor in the same hand and reduced arm swing while walking.
Examination reveals a pill-rolling tremor at rest, cogwheel rigidity, bradykinesia, and a stooped posture with shuffling gait.
The patient’s handwriting has become progressively smaller (micrographia), and he reports difficulty turning in bed and initiating movements.
These findings are consistent with idiopathic Parkinson’s disease (PD).
Epidemiology
Parkinson’s disease is the second most common neurodegenerative disorder after Alzheimer’s disease.
The global prevalence is approximately 100–200 per 100,000 population, increasing to over 1,000 per 100,000 in individuals over 80 years.
The mean age of onset is around 60 years, with a slight male predominance.
Both genetic and environmental factors contribute to disease risk, including pesticide exposure, rural living, and certain gene mutations (e.g., LRRK2, PARK7, SNCA).
Family history is present in 10–15% of patients, suggesting a multifactorial etiology with genetic susceptibility.
Etiopathophysiology
PD is characterized by progressive loss of dopaminergic neurons in the substantia nigra pars compacta of the midbrain. The resultant dopamine deficiency in the nigrostriatal pathway disrupts basal ganglia circuitry, leading to impaired motor control.
Histopathologically, Lewy bodies, composed primarily of aggregated α-synuclein, are the hallmark intracellular inclusions.
Oxidative stress, mitochondrial dysfunction, neuroinflammation, and impaired protein degradation are implicated in neuronal degeneration.
The imbalance between dopaminergic inhibition and cholinergic excitation in the basal ganglia underlies the cardinal motor symptoms.
Key Mechanisms: - Dopaminergic neuron loss in substantia nigra pars compacta (>60-80% loss by symptom onset) - Lewy body pathology (α-synuclein aggregation) - Mitochondrial dysfunction and oxidative stress - Impaired protein clearance (ubiquitin-proteasome system, autophagy) - Basal ganglia circuit disruption (direct and indirect pathways)
Clinical Features
Motor Symptoms (Cardinal Features): - The classic motor features include resting tremor (typically "pill-rolling"), bradykinesia, rigidity, and postural instability.
Tremor usually begins unilaterally, often in one hand, and diminishes with voluntary movement.
Bradykinesia manifests as reduced facial expression (hypomimia), soft speech (hypophonia), micrographia, and difficulty initiating or performing repetitive movements.
Gait abnormalities include reduced arm swing, shuffling steps, freezing episodes, and difficulty turning.
Non-Motor Symptoms (Often Precede Motor Signs): - Olfactory: Anosmia (loss of smell) - Gastrointestinal: Constipation, dysphagia - Sleep: REM sleep behavior disorder, insomnia - Psychiatric: Depression, anxiety, apathy - Autonomic: Orthostatic hypotension, urinary urgency - Cognitive: Executive dysfunction, later dementia (30-40%)
Asymmetric onset with resting tremor and clear levodopa responsiveness are hallmarks of idiopathic Parkinson’s disease. Non-motor symptoms (anosmia, REM sleep behavior disorder, constipation) often predate motor symptoms by years and are critical for early recognition.
Diagnosis and Differential Diagnosis
Clinical Diagnosis: - Diagnosis is clinical, based on the presence of cardinal motor features, asymmetry of symptoms, and response to dopaminergic therapy.
Supportive findings include resting tremor, progression over time, and levodopa responsiveness (>70% improvement).
Imaging with dopamine transporter SPECT (DaTscan) can support diagnosis by demonstrating reduced striatal uptake but is not routinely required.
Differential Diagnosis Comparison:
| Onset |
Asymmetric |
Symmetric |
Symmetric |
Symmetric |
| Tremor Type |
Resting (4-6 Hz) |
Rare/absent |
Rare/postural |
Action/postural (6-12 Hz) |
| Gait |
Shuffling, festination |
Early falls, backward |
Broad-based, early falls |
Normal |
| Eye Movements |
Normal |
Vertical gaze palsy |
Normal |
Normal |
| Autonomic |
Late, mild |
Mild |
Early, severe (OH, urinary) |
Absent |
| Levodopa Response |
Excellent (>70%) |
Poor (<30%) |
Poor/transient |
None |
| Cognitive |
Late dementia |
Early executive |
Rare |
Absent |
| Progression |
Slow (years) |
Rapid (3-5 yr) |
Rapid (5-10 yr) |
Stable |
Clinically Established PD requires: 1. Parkinsonism (bradykinesia + rest tremor OR rigidity) 2. Supportive Criteria (≥2): - Clear, dramatic response to dopaminergic therapy - Presence of levodopa-induced dyskinesia - Rest tremor in a limb - Olfactory loss or cardiac sympathetic denervation 3. No Absolute Exclusion Criteria: - Cerebellar signs, supranuclear gaze palsy - Frontotemporal dementia within 1 year - Parkinsonian features restricted to lower limbs for >3 years - Treatment with dopamine blockers - Absence of observable response to high-dose levodopa - Normal DaTscan 4. No Red Flags (see above)
Management
Pharmacologic Treatment (Motor Symptoms):
| Levodopa/DDI |
Carbidopa/levodopa |
25/100 mg TID, titrate to effect |
Dopamine precursor |
Grade A |
Gold standard; most effective; risk of dyskinesia with long-term use |
| Dopamine Agonists |
Pramipexole, ropinirole |
0.5-4.5 mg/day PO |
D2/D3 receptor agonist |
Grade A |
Delay motor complications; impulse control disorders risk |
| MAO-B Inhibitors |
Rasagiline, selegiline |
1 mg/day (rasagiline) |
Inhibit dopamine breakdown |
Grade A |
Mild symptomatic benefit; neuroprotection unclear |
| COMT Inhibitors |
Entacapone |
200 mg with each levodopa dose |
Prolong levodopa half-life |
Grade B |
Adjunct to levodopa; reduces “off” time |
| Anticholinergics |
Benztropine |
0.5-2 mg BID |
Reduce cholinergic excess |
Grade C |
For tremor; avoid in elderly (cognitive side effects) |
| Amantadine |
Amantadine |
100 mg BID-TID |
NMDA antagonist |
Grade B |
For dyskinesia; mild anti-parkinsonian effect |
Treatment Algorithm:
- Early PD (mild symptoms, age <65):
- First-line: Dopamine agonist OR MAO-B inhibitor [Grade A]
- Rationale: Delay levodopa, reduce dyskinesia risk
- Monitor for impulse control disorders with agonists
- Early PD (significant functional impairment OR age >65):
- First-line: Levodopa/carbidopa (start 25/100 mg TID) [Grade A]
- Rationale: Most effective, better tolerated in elderly
- Provides 4-6 years of good symptom control (“honeymoon period”)
- Moderate PD (motor fluctuations developing):
- Add: COMT inhibitor (entacapone) to reduce “off” time [Grade B]
- Add: Amantadine for emerging dyskinesia [Grade B]
- Add: MAO-B inhibitor if not already on it [Grade A]
- Advanced PD (severe motor fluctuations, dyskinesia):
- Consider: Deep brain stimulation (STN or GPi) [Grade A]
- Alternative: Levodopa/carbidopa intestinal gel (Duopa)
- Alternative: Apomorphine subcutaneous injections
Non-Motor Symptom Management:
| Depression |
SSRIs (citalopram, sertraline) |
10-20 mg/day |
Grade B |
| Psychosis |
Quetiapine, pimavanserin |
25-50 mg/day (quetiapine) |
Grade B |
| Orthostatic Hypotension |
Midodrine, fludrocortisone |
5-10 mg TID (midodrine) |
Grade C |
| Constipation |
Polyethylene glycol |
17 g/day |
Grade B |
| REM Sleep Disorder |
Melatonin, clonazepam |
3-12 mg HS (melatonin) |
Grade C |
| Dementia |
Rivastigmine |
4.6-9.5 mg/day patch |
Grade A |
| Drooling |
Glycopyrrolate, botulinum toxin |
1-2 mg TID (glycopyrrolate) |
Grade B |
Surgical/Device-Based Therapies: - Deep Brain Stimulation (DBS): STN or GPi target; reduces motor fluctuations by 50-70%, reduces medication needs by 30-50% [Grade A, Strong] - Indications: Motor fluctuations, medication-refractory tremor, disabling dyskinesia, good levodopa response, no significant cognitive impairment - Risks: Infection (3-5%), intracerebral hemorrhage (1-2%), cognitive/psychiatric effects
Non-Pharmacologic Interventions: - Physical therapy: Gait training, balance exercises, flexibility [Grade B] - Occupational therapy: ADL optimization, home safety [Grade B] - Speech therapy: For dysarthria, dysphagia [Grade C] - Exercise: Regular aerobic exercise improves motor function [Grade B]
Multiple Choice Question
Which of the following features most strongly supports a diagnosis of idiopathic Parkinson’s disease?
- Early postural instability within 6 months of onset
- Rapid progression with poor response to levodopa
- Symmetric onset of rigidity and tremor
- Asymmetric onset with clear levodopa responsiveness
Answer: (D) Asymmetric onset with clear levodopa responsiveness
References
- Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386(9996):896–912. DOI: 10.1016/S0140-6736(14)61393-3.
- Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, Obeso J, Marek K, Litvan I, Lang AE, Halliday G, Goetz CG, Gasser T, Dubois B, Chan P, Bloem BR, Adler CH, Deuschl G. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord. 2015;30(12):1591–1601. DOI: 10.1002/mds.26424.
- Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, Schrag AE, Lang AE. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013. DOI: 10.1038/nrdp.2017.13.
Peripheral Neuropathy
📊 Quick Facts
- Prevalence: 2–3% general population (up to 50% in diabetes)
- Patterns: Length-dependent distal symmetric > focal/multifocal
- Etiologies: Metabolic/toxic (~40%), immune/inflammatory, hereditary, infectious
- Autonomic Involvement: ~20% in diabetic neuropathy
- Impact: Leading cause of neuropathic pain and foot ulceration
⚠️ Red Flags
- Acute/subacute progression → Consider GBS, vasculitis, porphyria
- Asymmetry or multifocal deficits → Evaluate for mononeuritis multiplex
- Upper motor neuron signs → Central lesion (myelopathy)
- Prominent autonomic failure or weight loss → Amyloidosis, paraneoplastic
- Family history + early onset → Hereditary neuropathy (CMT, HNPP)
Clinical Scenario
A 58-year-old man with a 15-year history of poorly controlled type 2 diabetes presents with burning pain, numbness, and tingling in both feet for 8 months.
He reports difficulty walking in the dark and frequent tripping, though he denies muscle weakness.
Neurological examination reveals diminished vibration and pinprick sensation in a stocking-glove distribution, reduced ankle reflexes, and a positive Romberg sign.
Nerve conduction studies demonstrate reduced sensory nerve action potentials and slowed conduction velocities.
These findings are consistent with a length-dependent, symmetric, sensorimotor peripheral neuropathy, most likely diabetic in origin.
Epidemiology
Peripheral neuropathy refers to damage or dysfunction of peripheral nerves, affecting sensory, motor, or autonomic fibers.
It affects approximately 2–3% of the general population and up to 50% of individuals with long-standing diabetes.
The prevalence increases with age and is more common in males, particularly when related to metabolic or toxic etiologies.
Diabetes mellitus, chronic alcohol use, vitamin deficiencies, and autoimmune diseases are the most frequent causes.
Genetic causes (e.g., Charcot-Marie-Tooth disease) and paraneoplastic or infectious etiologies (e.g., HIV, leprosy) contribute to a smaller but significant proportion of cases.
Etiopathophysiology
- Axonal neuropathies arise from metabolic or toxic insults causing distal “dying-back” degeneration.
- Demyelinating neuropathies (CIDP, hereditary forms) show segmental myelin loss with slowed conduction and block.
- Small-fiber neuropathies injure thinly myelinated/unmyelinated fibers, leading to burning pain despite normal NCS; skin biopsy quantifies loss.
- Autonomic neuropathies reflect sympathetic/parasympathetic fiber loss, producing orthostasis, gastroparesis, and sexual dysfunction.
- Vasculitic/infiltrative processes cause focal ischemia with asymmetric deficits (mononeuritis multiplex).
Clinical Features
Sensory symptoms, including numbness, tingling, burning pain, and allodynia, often present in a "stocking-glove" distribution.
Motor involvement leads to distal weakness, muscle wasting, and reduced reflexes, while autonomic involvement can cause orthostatic hypotension, gastrointestinal dysmotility, and impotence.
Symptoms typically begin distally and ascend proximally over time (length-dependent pattern).
Focal mononeuropathies (e.g., carpal tunnel syndrome) and multifocal presentations (mononeuritis multiplex) may occur in vasculitic or infiltrative etiologies.
Severe cases may lead to gait instability, ulceration, and secondary infections due to sensory loss.
Length-dependent neuropathy marches “from toes to knees to fingers.” Ask about autonomic symptoms (orthostasis, gastroparesis) and inspect feet at every visit to prevent ulcers.
Diagnosis and Differential Diagnosis
Diagnosis involves a detailed history and neurological examination focusing on distribution, modality, and progression of symptoms.
Nerve conduction studies and electromyography (EMG) differentiate between axonal and demyelinating processes.
Laboratory workup includes glucose, HbA1c, B12, thyroid function, renal and liver function, and serum protein electrophoresis.
Nerve biopsy is reserved for suspected vasculitic, amyloid, or infiltrative neuropathies.
Differential diagnosis includes radiculopathy, myopathy, neuromuscular junction disorders, and central causes such as myelopathy or cortical lesions.
Differential Diagnosis Comparison:
| Time Course |
Chronic, gradual |
Subacute/chronic >8 wks |
Acute or stepwise asymmetric |
Acute/subacute |
| Distribution |
Stocking-glove |
Proximal + distal |
Multifocal (mononeuritis multiplex) |
Dermatomal |
| Motor vs Sensory |
Sensory > motor |
Motor + sensory |
Painful sensory/motor deficits |
Both within single root |
| Reflexes |
Reduced distally |
Globally reduced |
Reduced in affected nerves |
Reduced in root distribution |
| NCS/EMG |
Axonal loss |
Demyelinating features |
Axonal with conduction block |
Normal or root-level changes |
| Key Tests |
Metabolic labs, SPEP |
CSF protein, MRI roots |
ESR/CRP, ANCA, biopsy |
Spine MRI |
- History/exam: Onset, distribution, exposures, autonomic symptoms, family history.
- Screening labs (all): CBC, CMP, fasting glucose/HbA1c, B12 ± MMA, TSH, SPEP/UPEP with immunofixation.
- Second-line labs (as needed): ESR/CRP, ANA, ANCA, HIV, hepatitis, Lyme, heavy metals, genetic panels.
- Electrodiagnostics: NCS/EMG to classify axonal vs demyelinating, focal vs diffuse.
- Targeted tests: Skin biopsy for small fiber, nerve biopsy for vasculitis/amyloid, autonomic testing when dysautonomia suspected.
Management
Treat Underlying Etiology:
| Diabetes/metabolic |
Tight glycemic control, lifestyle modification, SGLT2/GLP-1 agents |
| Toxic/chemotherapy |
Dose reduction, alternative agents, antioxidant support |
| Autoimmune (CIDP, vasculitis) |
IVIG, corticosteroids, plasma exchange, rituximab |
| Nutritional deficiency |
Vitamin repletion (B12, thiamine) |
| Infectious |
Pathogen-specific therapy (HIV, Lyme, leprosy) |
| Hereditary |
Genetic counseling, supportive care |
Neuropathic Pain Control:
| Gabapentinoids |
Gabapentin, pregabalin |
Grade A |
First-line for painful diabetic neuropathy |
| SNRIs |
Duloxetine, venlafaxine |
Grade A |
Benefit mood disorders |
| TCAs |
Amitriptyline, nortriptyline |
Grade B |
Use bedtime dosing; caution in elderly |
| Topicals |
Lidocaine 5% patch, capsaicin 8% |
Grade B |
Focal neuropathic pain |
| Opioids/tramadol |
Second/third-line |
Grade C |
Short-term rescue only |
Supportive & Preventive Care: - Foot care/podiatry: Daily inspection, custom footwear, ulcer prevention. - Physical/occupational therapy: Balance training, gait aids, orthoses for foot drop. - Autonomic management: Compression stockings, midodrine/fludrocortisone for orthostasis; dietary changes for gastroparesis, bladder programs. - Education: Fall prevention, avoid barefoot walking, temperature checks to prevent burns.
Management Algorithm: 1. Classify neuropathy pattern (distal symmetric vs focal/multifocal; sensory vs motor). 2. Perform screening labs + NCS/EMG; pursue targeted tests based on findings. 3. Address reversible causes (optimize diabetes, stop neurotoxic agents, treat infections). 4. Initiate pain therapy (gabapentinoid or SNRI) and titrate; add topical agents as needed. 5. Escalate for immune-mediated disease with IVIG, steroids, or plasma exchange; consider rituximab/cyclophosphamide for refractory vasculitis. 6. Long-term follow-up: Monitor for progression, reinforce foot care, adjust therapy, and screen for autonomic or motor complications.
Multiple Choice Question
Which of the following features most strongly suggests a demyelinating rather than axonal peripheral neuropathy?
- Length-dependent distal sensory loss
- Significantly slowed nerve conduction velocity
- Muscle wasting and fasciculations
- Reduced compound motor action potential amplitude
Answer: (B) Significantly slowed nerve conduction velocity
References
- Dyck PJ, Thomas PK (eds). Peripheral Neuropathy, 5th ed. Elsevier,
- England JD, Gronseth GS, Franklin G, et al. Practice parameter: Evaluation of distal symmetric polyneuropathy: Role of laboratory and genetic testing (an evidence-based review). Neurology. 2009;72(2):185–192. DOI: 10.1212/01.wnl.0000336345.70511.0f.
- Said G. Diabetic neuropathy — a review. Nat Clin Pract Neurol. 2007;3(6):331–340. DOI: 10.1038/ncpneuro0504.
Post-Concussion Syndrome
📊 Quick Facts
- Incidence: 10–30% of mild TBI patients
- Risk Factors: Prior concussion, psychiatric history, female sex, older age
- Symptoms: Somatic, cognitive, emotional clusters lasting >2–4 weeks
- Course: Majority recover by 3 months, but subset >1 year
- Impact: Major driver of time off work/school after concussion
⚠️ Red Flags
- Progressive neurological deficits → evaluate for structural lesion
- Persistent vomiting, severe worsening headache → repeat imaging
- Focal seizures or focal deficits → rule out intracranial pathology
- Marked mood changes with suicidality → urgent psychiatric evaluation
- Ocular or vestibular symptoms impairing safety → refer to specialist
Clinical Scenario
A 34-year-old man presents 6 weeks after a mild traumatic brain injury sustained in a bicycle accident, during which he did not lose consciousness but was briefly disoriented.
He reports persistent headaches, difficulty concentrating, memory lapses, and increased irritability interfering with his work.
He also describes sleep disturbances, fatigue, and heightened sensitivity to light and noise.
Neurological examination is normal, and routine imaging shows no acute abnormalities.
His presentation is consistent with post-concussion syndrome (PCS), a common but often under-recognized sequela of mild traumatic brain injury.
Epidemiology
Post-concussion syndrome occurs in approximately 10–30% of individuals following mild traumatic brain injury (mTBI), though prevalence varies across studies.
It is more common in adults than children and occurs more frequently in females and older patients.
Risk factors include prior concussions, pre-existing psychiatric conditions, and lower educational attainment.
Most patients develop symptoms within days to weeks after injury, and while many recover within 3 months, a significant minority experience symptoms for a year or more.
PCS contributes substantially to healthcare utilization and socioeconomic burden due to prolonged cognitive and functional impairment.
Etiopathophysiology
- Mild TBI produces transient ionic shifts, axonal stretching, and altered cerebral metabolism (“energy crisis”).
- Persistent symptoms may reflect diffuse axonal injury, impaired autonomic regulation, neuroinflammation, and neurotransmitter imbalance.
- Functional MRI/PET show network connectivity changes even when structural imaging is normal.
- Psychological factors (anxiety, depression, PTSD) interact with biological injury to perpetuate symptoms.
Clinical Features
PCS manifests as a constellation of somatic, cognitive, and emotional symptoms that persist beyond the expected recovery period of a concussion.
Somatic features include headache, dizziness, photophobia, phonophobia, and sleep disturbances.
Cognitive symptoms include difficulty concentrating, memory impairment, slowed processing speed, and reduced executive function.
Emotional and behavioral manifestations such as irritability, anxiety, depression, and emotional lability are common.
Symptoms often fluctuate and may be exacerbated by stress, fatigue, or additional head trauma.
Reassure patients that gradual return to activity is safe—prolonged complete rest (>48 hours) may actually worsen recovery; target symptom-limited physical and cognitive loading.
Diagnosis and Differential Diagnosis
PCS is a clinical diagnosis, established when symptoms persist for weeks to months following a concussion and cannot be explained by other causes.
Neuroimaging is usually normal but may be used to exclude structural lesions or other intracranial pathology.
Neuropsychological testing can help characterize cognitive deficits and guide rehabilitation strategies.
Differential diagnoses include chronic subdural hematoma, depression, post-traumatic stress disorder, vestibular dysfunction, and migraine.
It is essential to distinguish PCS from malingering or symptom exaggeration, particularly in medico-legal contexts.
Differential Diagnosis Comparison:
| Onset after injury |
Days–weeks, persistent >4 wks |
Weeks–months with progressive deficits |
Variable, trauma-related |
Immediate dizziness/imbalance |
| Imaging |
Normal |
Subdural collection on CT/MRI |
Normal |
Normal/vestibular testing abnormal |
| Symptoms |
Headache, cognitive fog, mood changes |
Focal deficits, gait disturbance |
Hyperarousal, reliving trauma |
Vertigo, imbalance |
| Exam |
Often normal |
Focal neuro signs |
Psychiatric signs |
Abnormal vestibular tests |
- Comprehensive history/exam focusing on symptom clusters, triggers, and red flags.
- Screening tools: Post-Concussion Symptom Scale, SCAT-6, mood/anxiety questionnaires.
- Neuropsychological testing for persistent cognitive complaints.
- Vestibular/oculomotor assessment in patients with dizziness or visual strain.
- Imaging/labs only when atypical features or deterioration warrants further evaluation.
Management
Foundational Strategies:
| Education & reassurance |
Normalize recovery trajectory, set expectations |
| Graded return to activity |
Symptom-limited cognitive/physical rest for 24–48 h, then progressive loading |
| Sleep hygiene & lifestyle |
Regular sleep schedule, hydration, nutrition, limit alcohol/caffeine |
Symptom-Targeted Therapies:
| Headache |
Non-opioid analgesics, migraine prophylaxis (topiramate, amitriptyline), nerve blocks for occipital headache |
| Cognitive slowing |
Cognitive rehab, occupational therapy, structured routines |
| Mood/Anxiety |
SSRIs/SNRIs, CBT, mindfulness therapies |
| Sleep disturbances |
Sleep hygiene, short-term melatonin, CBT for insomnia |
| Vestibular/ocular symptoms |
Vestibular rehab, vision therapy, prism glasses |
| Autonomic dysregulation |
Hydration, graded exercise, consider fludrocortisone or beta-blockers in selected cases |
Management Algorithm: 1. Initial assessment (first 1–2 weeks): Rule out red flags, provide education, brief rest, plan gradual return. 2. Persistent symptoms (>4 weeks): Multidisciplinary evaluation (neuropsychology, vestibular, mental health). 3. Targeted treatments for dominant symptom clusters; address comorbidities (migraine, mood disorders, sleep apnea). 4. Monitor progress every 2–4 weeks; escalate to specialty concussion clinic if refractory. 5. Vocational/school accommodations and social support for prolonged cases.
Multiple Choice Question
Which of the following best describes post-concussion syndrome (PCS)?
- Structural brain injury with progressive neurological deterioration following traumatic brain injury
- A chronic neurodegenerative condition characterized by abnormal protein aggregation
- A constellation of cognitive, emotional, and somatic symptoms
- persisting beyond the typical recovery period after a mild traumatic
Answer: (C) A constellation of cognitive, emotional, and somatic symptoms
References
- McCrory P, Meeuwisse W, Dvořák J, et al. Consensus statement on concussion in sport—the 5th International Conference on Concussion in Sport held in Berlin, October 2016. Br J Sports Med. 2017;51(11):838–847. DOI: 10.1136/bjsports-2017-097699
- Silverberg ND, Iverson GL. Is rest after concussion “the best medicine?”: recommendations for activity resumption following concussion in athletes, civilians, and military service members. J Head Trauma Rehabil. 2013;28(4):250–259. DOI: 10.1097/HTR.0b013e31825ad658
- Renga V. Clinical evaluation of patients with vestibular dysfunction. Neurol Res Int. 2019;2019:3931548. DOI: 10.1155/2019/3931548 PMID: 30863640 PMCID: PMC6377969
Posterior Cortical Atrophy

Clinical Scenario
A 62-year-old woman presents with progressive difficulty reading, judging distances, and recognizing faces over the past two years.
She reports no significant memory loss but struggles with dressing and navigating familiar environments.
Neurological examination reveals visuospatial deficits, optic ataxia, and elements of Balint syndrome, while language and motor function remain intact.
MRI of the brain shows prominent parietal and occipital cortical atrophy, with relative sparing of the hippocampi.
These findings suggest posterior cortical atrophy (PCA), a neurodegenerative syndrome primarily affecting posterior cortical networks.
Epidemiology
PCA is a rare, early-onset neurodegenerative condition most commonly linked to Alzheimer’s disease (AD), accounting for 5% of AD cases.
The mean age of onset is typically between 50 and 65 years, and the disease affects men and women equally.
Due to its rarity and atypical presentation, PCA is often misdiagnosed as primary ophthalmologic or psychiatric disease.
Most cases are sporadic, but rare familial forms associated with APP, PSEN1, or PSEN2 mutations have been reported.
The average disease duration is approximately 8–12 years, similar to typical AD, but patients often retain memory until late stages.
Etiopathophysiology
PCA results from selective neurodegeneration in the parieto-occipital and posterior temporal cortices.
The majority of cases (≈80%) are due to underlying Alzheimer’s pathology with amyloid-β plaques and neurofibrillary tangles.
Less commonly, PCA can result from dementia with Lewy bodies, corticobasal degeneration, or prion disease.
The selective vulnerability of posterior cortical regions leads to prominent visuospatial and visuoperceptual dysfunction.
Neuroimaging and neuropathology show marked atrophy and hypometabolism in dorsal and ventral visual processing streams.
Clinical Features
Patients typically present with progressive visual processing deficits despite preserved visual acuity.
Key features include visuospatial disorientation, simultanagnosia, optic ataxia, ocular apraxia, and visual agnosia.
Gerstmann syndrome (acalculia, agraphia, left-right disorientation, finger agnosia) and alexia without agraphia may also occur.
Unlike typical Alzheimer’s disease, episodic memory, insight, and language are relatively spared in early stages.
As the disease progresses, cognitive decline becomes more global, and features of typical Alzheimer’s dementia emerge.
Diagnosis and Differential Diagnosis
Diagnosis is based on progressive visuospatial and visuoperceptual dysfunction with posterior cortical involvement on neuroimaging.
MRI shows parieto-occipital atrophy, and FDG-PET or SPECT demonstrates hypometabolism in posterior cortices.
Cerebrospinal fluid biomarkers (low Aβ42, elevated tau) or amyloid PET may support underlying Alzheimer’s pathology.
Differential diagnoses include dementia with Lewy bodies (visual hallucinations, parkinsonism), corticobasal syndrome, and Creutzfeldt-Jakob disease.
Ophthalmologic conditions (e.g., macular degeneration, glaucoma) should be excluded before diagnosing PCA.
Management
No disease-modifying therapies exist; management focuses on symptomatic treatment and functional support.
Cholinesterase inhibitors (e.g., donepezil) or memantine may provide cognitive benefit, particularly in Alzheimer’s-related PCA.
Occupational therapy, visual rehabilitation, and adaptive strategies (e.g., high-contrast environments, large-print materials) are crucial.
Psychological support for patients and caregivers is essential due to high rates of anxiety and depression.
Emerging disease-modifying approaches, including anti-amyloid monoclonal antibodies, are under investigation for underlying AD pathology.
Multiple Choice Question
Which of the following features most strongly supports a diagnosis of posterior cortical atrophy over typical Alzheimer’s disease?
- Early impairment of episodic memory
- Prominent visuospatial dysfunction with preserved visual acuity
- Early aphasia and apraxia of speech
- Parkinsonism and visual hallucinations
Answer: (B) Prominent visuospatial dysfunction with preserved visual acuity
References
- Crutch SJ, Schott JM, Rabinovici GD, et al. Consensus classification of posterior cortical atrophy. Alzheimers Dement. 2017;13(8):870–884. DOI: 10.1016/j.jalz.2017.01.014
- Tang-Wai DF, Graff-Radford NR, Boeve BF, et al. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology. 2004;63(7):1168–1174. DOI: 10.1212/01.WNL.0000140289.18472.15
- Renner JA, Burns JM, Hou CE, et al. Progressive posterior cortical dysfunction: a clinicopathologic series. Neurology. 2004;63(7):1175–1180. DOI: 10.1212/01.wnl.0000140290.80962.bf
Posterior Interosseous Nerve Syndrome

Clinical Scenario
A 48-year-old carpenter presents with progressive weakness of finger extension over the past 3 weeks.
He denies pain, numbness, or tingling in the hand but reports difficulty in extending his fingers and wrist when using tools.
On examination, there is marked weakness of finger and thumb extension, but wrist extension is partially preserved due to intact extensor carpi radialis longus function.
Sensory examination is normal, and Tinel’s sign is negative over the carpal tunnel.
These findings suggest a diagnosis of posterior interosseous nerve (PIN) syndrome.
Epidemiology
PIN syndrome is an uncommon compressive neuropathy of the deep branch of the radial nerve, typically occurring in adults aged 40–60 years.
It is more frequent in individuals engaged in repetitive forearm pronation-supination activities, such as manual laborers, athletes, and musicians.
It accounts for less than 0.7% of all upper extremity nerve entrapments.
The condition shows no significant sex predilection.
Most cases are unilateral, although bilateral involvement can occur rarely, particularly in systemic neuropathies.
Etiopathophysiology
The posterior interosseous nerve is a purely motor branch of the radial nerve that originates near the lateral epicondyle and passes through the supinator muscle (Arcade of Frohse).
Compression most commonly occurs at the Arcade of Frohse, but may also occur at the radial tunnel, fibrous bands, or vascular leash of Henry.
Traction injury, space-occupying lesions (e.g., lipomas, ganglion cysts), or iatrogenic injury during orthopedic procedures can also cause PIN palsy.
Entrapment leads to demyelination and axonal degeneration, resulting in impaired conduction to extensor muscles.
Chronic compression may lead to irreversible denervation if not addressed promptly.
Clinical Features
The hallmark is motor weakness without sensory deficit, as the PIN carries no cutaneous sensory fibers.
Patients exhibit weakness or paralysis of finger and thumb extensors, often presenting with a characteristic "finger drop."
Wrist extension is usually preserved but may show radial deviation due to intact extensor carpi radialis longus.
Pain is typically minimal or absent, distinguishing PIN syndrome from radial tunnel syndrome, which is predominantly painful.
Muscle wasting may develop in chronic cases, particularly in the extensor digitorum communis and extensor pollicis longus.
Diagnosis and Differential Diagnosis
Diagnosis is based on clinical findings of painless motor weakness with preserved sensation.
Electromyography (EMG) and nerve conduction studies (NCS) confirm denervation in PIN-innervated muscles and help localize the lesion.
MRI or ultrasound can identify structural causes such as tumors, ganglion cysts, or muscle hypertrophy compressing the nerve.
Differential diagnoses include cervical radiculopathy (C7-C8), radial nerve palsy at the spiral groove, motor neuron disease, and tendon rupture.
Exclusion of central causes (e.g., stroke) is essential if upper motor neuron signs are present.
Management
Initial management involves activity modification, splinting, and nonsteroidal anti-inflammatory drugs (NSAIDs) to reduce inflammation.
Physical and occupational therapy are beneficial for preventing contractures and maintaining joint mobility.
If compression is due to a mass or if there is no improvement after 3–6 months of conservative therapy, surgical decompression is indicated.
Postoperative prognosis is generally good, with functional recovery in 80–90% of cases if intervention occurs before significant axonal loss.
Early diagnosis and treatment are crucial for preventing permanent motor deficits.
Multiple Choice Question
Which of the following clinical findings best distinguishes posterior interosseous nerve syndrome from radial tunnel syndrome?
- Pain in the lateral forearm during pronation
- Paresthesia over the dorsum of the hand
- Motor weakness of finger extension without sensory deficit
- Tenderness over the lateral epicondyle
Answer: (C) Motor weakness of finger extension without sensory deficit
References
- Spinner M. Injuries to the Major Branches of Peripheral Nerves of the Forearm. 2nd ed. Philadelphia: W.B. Saunders; 1978.
- Roles NC, Maudsley RH. Radial tunnel syndrome: resistant tennis elbow as a nerve entrapment. J Bone Joint Surg Br. 1972;54(3):499–508. DOI: 10.1302/0301-620X.54B3.499
Posterior Reversible Encephalopathy Syndrome (PRES)

📊 Quick Facts
- Syndrome: Neurotoxic, usually reversible
- Triggers: Severe hypertension, renal failure, eclampsia, cytotoxic/biologic therapy, autoimmune disease
- Presentation: Seizures, encephalopathy, visual changes, headache
- Imaging: Posterior vasogenic edema, but atypical locations possible
- Outcome: Reversible in days–weeks with prompt treatment; recurrence uncommon but possible
⚠️ Red Flags
- Hypertensive emergency with neurological symptoms
- Pregnant/postpartum patient with seizures or vision loss
- Immunosuppressed patient on calcineurin inhibitors
- Refractory seizures/status epilepticus
- Signs of intracranial hypertension or hemorrhage
Clinical Scenario
A 54-year-old man with a history of uncontrolled hypertension, poorly compliant with medications, presents with a new-onset generalized tonic-clonic seizure.
On arrival, he is confused and drowsy, with a blood pressure of 220/120 mmHg.
Neurological examination shows no focal motor deficits, but transient visual blurring is reported.
MRI brain (Axial FLAIR) demonstrates symmetric hyperintense lesions in the parieto-occipital white matter, consistent with Posterior Reversible Encephalopathy Syndrome (PRES).
Blood pressure control and supportive therapy are initiated, leading to significant clinical improvement over the next few days.
Epidemiology
PRES is a neurotoxic syndrome characterized by acute neurological symptoms and distinctive radiological findings, typically reversible with prompt treatment.
It can occur in both adults and children, with a higher prevalence in women, particularly in the context of autoimmune diseases or preeclampsia/eclampsia.
The incidence is increasing due to heightened clinical recognition and widespread use of MRI.
Common predisposing conditions include acute hypertension, renal failure, cytotoxic or immunosuppressive therapy, and autoimmune diseases.
Although often reversible, delayed recognition or inadequate management may lead to permanent neurological deficits or death.
Etiopathophysiology
- Failure of cerebral autoregulation from abrupt hypertension causes hyperperfusion, blood–brain barrier disruption, and vasogenic edema.
- Endothelial injury from toxins (calcineurin inhibitors, chemotherapy), autoimmune disease, or sepsis increases vascular permeability even without severe hypertension.
- Posterior circulation is more susceptible due to sparse sympathetic innervation, explaining parieto-occipital predominance.
- Severe cases can develop cytotoxic edema, hemorrhage, or infarction.
Clinical Features
PRES presents acutely or subacutely, often within hours to days, with a range of neurological symptoms.
Seizures occur in up to 70–90% of cases and are often the presenting feature.
Other common manifestations include headache, encephalopathy, visual disturbances (e.g., blurred vision, cortical blindness), and nausea/vomiting.
Focal neurological deficits such as hemiparesis or aphasia are less common but may occur.
Symptoms are usually reversible with prompt recognition and treatment but may progress to coma or status epilepticus if untreated.
Diagnosis and Differential Diagnosis
Diagnosis is primarily based on clinical presentation and characteristic neuroimaging findings.
MRI typically shows bilateral, symmetric T2/FLAIR hyperintensities in the parieto-occipital lobes, representing vasogenic edema.
Atypical patterns involving the frontal lobes, basal ganglia, brainstem, or cerebellum may also occur.
Differential diagnoses include ischemic or hemorrhagic stroke, cerebral venous thrombosis, infectious or autoimmune encephalitis, reversible cerebral vasoconstriction syndrome (RCVS), and demyelinating diseases.
CSF analysis and EEG may assist in excluding other causes, especially in atypical cases.
Differential Diagnosis Comparison:
| Onset |
Acute/subacute |
Acute with malignant HTN |
Thunderclap headaches |
Subacute |
| BP |
Often elevated but not always |
Markedly elevated |
Variable |
Variable |
| Imaging |
Posterior vasogenic edema |
Diffuse edema |
Multifocal arterial narrowing, infarcts |
Limbic/cortical signal change |
| CSF |
Usually normal/slight protein ↑ |
Normal |
Mild pleocytosis |
Lymphocytic pleocytosis |
| Treatment |
Remove trigger, control BP |
Aggressive BP lowering |
Calcium channel blockers, treat triggers |
Immunotherapy |
- Clinical suspicion in patients with seizures, headache, vision changes + risk factors.
- MRI brain with FLAIR/DWI to identify vasogenic edema; consider gadolinium for atypical patterns.
- Vascular imaging (MRA/CTA) to exclude RCVS or arterial occlusion; MRV if CVST suspected.
- Laboratory evaluation: CBC, CMP, renal function, autoimmune markers, drug/toxin levels.
- CSF/EEG when infection or autoimmune encephalitis cannot be excluded.
Management
Immediate Actions:
| Remove trigger |
Control hypertension, stop offending drugs (calcineurin inhibitors, chemo), treat preeclampsia/eclampsia |
| Stabilize |
ICU monitoring, airway protection if seizures/status epilepticus |
| Blood pressure control |
Reduce MAP by 20–25% over first few hours using IV agents (nicardipine, labetalol); avoid rapid overcorrection |
| Seizure control |
Benzodiazepines, IV antiseizure meds; discontinue once PRES resolves unless recurrence |
Supportive & Follow-Up Care: - Manage intracranial pressure if needed (head elevation, hyperosmolar therapy). - Treat renal failure, autoimmune disease, or sepsis contributing to endothelial injury. - Repeat MRI in 1–2 weeks to confirm resolution; counsel on recurrence risk.
Management Algorithm: 1. Recognize syndrome in high-risk patients with acute neurological symptoms. 2. Initiate blood pressure control and remove precipitating agents. 3. Treat seizures/status epilepticus; consider continuous EEG. 4. Supportive care with ICU monitoring, manage complications (edema, hemorrhage). 5. Long-term strategies: optimize BP, renal function, and adjust immunosuppressive regimens to prevent recurrence.
Multiple Choice Question
Which of the following MRI findings is most characteristic of PRES?
- Restricted diffusion in the basal ganglia with hemorrhage
- Bilateral parieto-occipital vasogenic edema on T2/FLAIR
- Diffuse leptomeningeal enhancement with mass effect
- Unilateral cortical infarction in the MCA territory
Answer: (B) Bilateral parieto-occipital vasogenic edema on T2/FLAIR PRES
References
- Fugate JE, Rabinstein AA. Posterior reversible encephalopathy syndrome: clinical and radiological manifestations, pathophysiology, and outstanding questions. Lancet Neurol. 2015;14(9):914–925. DOI: 10.1016/S1474-4422(15)00111-8
- Bartynski WS. Posterior reversible encephalopathy syndrome, Part 1: fundamental imaging and clinical features. AJNR Am J Neuroradiol. 2008;29(6):1036–1042. DOI: 10.3174/ajnr.A0928
- Lee VH, Wijdicks EF, Manno EM, Rabinstein AA. Clinical spectrum of reversible posterior leukoencephalopathy syndrome. Arch Neurol. 2008;65(2):205–210. DOI: 10.1001/archneurol.2007.46
Postural Orthostatic Tachycardia Syndrome (POTS)

- Prevalence: 0.2-1% of general population; often underdiagnosed
- Demographics: Young women (15-45 years); F:M ratio 5:1
- Diagnostic criterion: HR increase >30 bpm within 10 min of standing (no BP drop)
- Classic symptoms: Lightheadedness, palpitations, fatigue, “brain fog”
- First-line treatment: Volume expansion (fluids + salt), compression, exercise
- Prognosis: Often improves with treatment; rarely progresses
- Syncope with standing - suggest additional evaluation for cardiac causes
- Chest pain or dyspnea - rule out cardiac/pulmonary pathology
- New onset in older adults - consider secondary causes (anemia, dehydration)
- Progressive neurological deficits - exclude primary autonomic failure
- Severe weight loss - investigate for underlying systemic disease
- Supine hypertension - may develop with treatment; adjust medications
- Post-viral onset - increasingly recognized (including post-COVID)
Clinical Scenario
A 24-year-old woman presents with episodes of lightheadedness, palpitations, and near-syncope upon standing that improve when lying down. She reports significant fatigue, exercise intolerance, and occasional "brain fog" affecting her concentration. Physical examination is unremarkable except for marked increase in heart rate from 75 bpm supine to 120 bpm within 8 minutes of standing, without significant orthostatic hypotension. Laboratory workup including thyroid function, electrolytes, and CBC is normal. Tilt-table testing confirms a diagnosis of postural orthostatic tachycardia syndrome (POTS).
Epidemiology
- Chronic orthostatic intolerance with excessive tachycardia upon standing (no significant BP drop)
- Prevalence: 0.2-1% of general population; likely underdiagnosed
- Predominantly affects young women aged 15-45 years; F:M ratio ~5:1
- Many cases idiopathic; may develop post-viral infection, surgery, pregnancy, or trauma
- Associated conditions: Ehlers-Danlos syndrome, autoimmune diseases, chronic fatigue syndrome, mast cell disorders
Autonomic dysregulation: - Heterogeneous condition with autonomic nervous system dysfunction - Exaggerated sympathetic activity upon standing - Impaired vasoconstriction → excessive venous pooling → compensatory tachycardia - Goal: maintain cerebral perfusion despite orthostatic stress
Contributing mechanisms: - Hypovolemia (reduced plasma volume) - Reduced plasma norepinephrine clearance - Abnormal baroreflex sensitivity - Autoimmune mechanisms (antibodies against adrenergic/muscarinic receptors in some)
POTS subtypes:
- Neuropathic POTS:
- Peripheral sympathetic denervation (especially lower extremities)
- Impaired vasoconstriction → venous pooling
- Normal or low plasma norepinephrine
- Hyperadrenergic POTS:
- Excessive sympathetic tone
- Elevated plasma norepinephrine (standing >600 pg/mL)
- May have supine hypertension, tremor, anxiety
- Hypovolemic POTS:
- Low blood volume (up to 30% reduction)
- Inadequate venous return
- Often responds to volume expansion
Clinical Features
Cardinal feature: - Orthostatic tachycardia without hypotension (distinguishes from orthostatic hypotension)
Orthostatic symptoms (occur upon standing): - Palpitations, lightheadedness, presyncope - Fatigue, weakness - Tremulousness - Cognitive difficulties ("brain fog") - Visual disturbances (blurred vision, tunnel vision)
Non-orthostatic symptoms: - Exercise intolerance - Headaches (often migraine-like) - Nausea, abdominal pain - Gastrointestinal dysmotility - Sleep disturbances
Physical findings: - Dependent acrocyanosis (bluish discoloration of feet/legs when standing) - Peripheral venous pooling - Livedoreticularis in some cases - Rarely true syncope (presyncope more common)
Impact: - Chronic, debilitating symptoms - Significant impact on quality of life, work, school performance
Diagnosis
Diagnostic criteria: - Sustained HR increase ≥30 bpm within 10 minutes of standing (≥40 bpm in adolescents 12-19 years) - No orthostatic hypotension (BP drop <20/10 mmHg) - Symptoms associated with standing - Duration ≥3 months
Diagnostic testing: - Active standing test (bedside): measure HR and BP supine, then at 2, 5, 10 min standing - Tilt-table testing (formal): 70° tilt for up to 30-45 minutes - Continuous HR and BP monitoring during test
Subtyping tests: - Plasma catecholamines (supine and standing): hyperadrenergic if standing NE >600 pg/mL - Autonomic reflex testing: assess for peripheral denervation - Blood volume assessment: identify hypovolemia
Exclude secondary causes: - Labs: CBC (anemia), TSH (hyperthyroidism), glucose, electrolytes - Medication review: diuretics, vasodilators, stimulants - Dehydration, deconditioning
Screen for associated conditions: - Ehlers-Danlos syndrome (joint hypermobility) - Autoimmune disorders - Mast cell activation syndrome - Small fiber neuropathy
Differential diagnosis: - Inappropriate sinus tachycardia - Orthostatic hypotension - Vasovagal syncope - Anxiety disorder with hyperventilation - Pheochromocytoma (if hyperadrenergic features)
Management
Treatment is individualized based on subtype and symptom severity. Non-pharmacologic measures are first-line for all patients.
Non-Pharmacologic Strategies (First-Line):
| Volume expansion |
2–3 L fluid/day + 3–10 g sodium/day |
| Compression |
Waist-high compression stockings or abdominal binders |
| Exercise |
Graduated recumbent/aquatic training transitioning to upright exercise |
| Lifestyle |
Avoid prolonged standing, heat, large meals; elevate head of bed |
Pharmacologic Options (Tailored to Subtype):
| Hypovolemia |
Fludrocortisone |
Expands plasma volume; monitor BP/K+ |
| Venous pooling |
Midodrine, droxidopa |
Vasoconstrictors; avoid supine hypertension |
| Tachycardia |
Low-dose propranolol, ivabradine |
Reduce HR; ivabradine useful when β-blockers contraindicated |
| Hyperadrenergic |
Clonidine, methyldopa |
Reduce sympathetic tone |
| Neuropathic |
Pyridostigmine |
Enhances ganglionic transmission |
Supportive Care: - Multidisciplinary care (autonomic specialist, PT, psychology). - Treat comorbid migraines, sleep disorders, mood disorders. - Consider CBT for coping; encourage hydration strategies during illness.
Management Algorithm: 1. Confirm diagnosis, exclude mimics, identify subtype. 2. Initiate lifestyle/volume measures with compression and exercise. 3. Add pharmacotherapy based on symptom profile (start low, titrate). 4. Address comorbidities and provide psychological support. 5. Reassess regularly; adjust therapy, consider autonomic referral or specialized rehab if refractory.
Multiple Choice Question
Which of the following findings is most characteristic of POTS?
- A sustained drop in systolic blood pressure of >20 mmHg within 3 minutes of standing
- A sustained heart rate increase of >30 bpm within 10 minutes of standing without orthostatic hypotension
- Sinus bradycardia during tilt-table testing
- Absence of sympathetic activation upon standing
Answer: (B) A sustained heart rate increase of more than 30 bpm within 10 minutes of standing without orthostatic hypotension.
References
- Raj SR. Postural tachycardia syndrome (POTS). Circulation. 2013;127(23):2336–2342. DOI: 10.1161/CIRCULATIONAHA.112.144501
- Vernino S, Bourne KM, Stiles LE, Grubb BP, Fedorowski A, Stewart JM, Arnold AC, Pace LA, Axelsson J, Boris JR, Moak JP, Goodman BP, Chémali KR, Chung TH, Goldstein DS, Diedrich A, Miglis MG, Cortez MM, Miller AJ, Freeman R, Biaggioni I, Rowe PC, Sheldon RS, Shibao CA, Systrom DM, Cook GA, Doherty TA, Abdallah HI, Darbari A, Raj SR. Postural orthostatic tachycardia syndrome (POTS): State of the science and clinical care from a 2019 National Institutes of Health Expert Consensus Meeting – Part 1. Auton Neurosci. 2021;235:102828. DOI: 10.1016/j.autneu.2021.102828 PMID: 34144933. PMCID: PMC8455420
- Sheldon RS, Grubb BP II, Olshansky B, et al. Heart Rhythm Society expert consensus statement on the diagnosis and treatment of postural tachycardia syndrome, inappropriate sinus tachycardia, and vasovagal syncope. Heart Rhythm. 2015;12(6):e41–e63. DOI: 10.1016/j.hrthm.2015.03.029
Sarcoidosis of Nervous system (Neurosarcoidosis)
- Incidence: 5-15% of systemic sarcoidosis patients develop neurosarcoidosis
- Peak age: 20-50 years; more common in women, African Americans
- Classic presentation: Cranial neuropathies (facial nerve palsy most common)
- Key imaging: Leptomeningeal enhancement on MRI with contrast
- Histology: Non-caseating granulomas (gold standard)
- Treatment: High-dose corticosteroids ± immunosuppressants
- Bilateral facial nerve palsy - highly suggestive of neurosarcoidosis
- Cranial neuropathies with systemic symptoms (cough, dyspnea, uveitis)
- Chronic meningitis unresponsive to antibiotics
- Diabetes insipidus - hypothalamic-pituitary involvement
- Rapidly progressive symptoms - aggressive disease requiring urgent treatment
- Hydrocephalus from chronic meningeal inflammation
- Unexplained seizures with leptomeningeal enhancement
Clinical Scenario
A 45-year-old African American woman presents with progressive facial weakness, intermittent diplopia, and chronic headaches over 3 months. She reports fatigue, weight loss, and persistent dry cough. Neurological examination reveals bilateral facial nerve palsy and mild sensory loss in the left leg. MRI brain shows leptomeningeal enhancement, and CSF analysis reveals lymphocytic pleocytosis with elevated protein. Biopsy of mediastinal lymph nodes demonstrates non-caseating granulomas, confirming the diagnosis of neurosarcoidosis.
Epidemiology
- Systemic sarcoidosis affects 10-20 per 100,000 individuals worldwide
- Neurosarcoidosis occurs in 5-15% of systemic sarcoidosis patients; may be initial or sole manifestation
- Peak age 20-50 years; female predominance; more common in African Americans and Northern Europeans
- Often underdiagnosed due to varied presentations mimicking other inflammatory/neoplastic disorders
- Higher mortality than systemic disease alone due to vital structure involvement
Immune dysregulation: - Dysregulated immune response to unidentified antigen → non-caseating granuloma formation - CD4+ T-helper cells and macrophages accumulate in affected tissues - Cytokine release (IL-2, TNF-α) sustains chronic inflammation
Nervous system involvement: - Granulomas infiltrate meninges, cranial nerves, hypothalamus, pituitary, or parenchyma - Blood-brain barrier disruption → perivascular inflammation - Tissue injury and demyelination result from chronic inflammation
Risk factors: - Genetic predisposition (HLA-DRB1 alleles) - Environmental exposures influence susceptibility and severity
Patterns of involvement: - Leptomeningeal (most common) - cranial nerves, chronic meningitis - Parenchymal - brain/spinal cord lesions mimicking MS - Hypothalamic-pituitary - endocrine dysfunction - Peripheral nervous system - mononeuritis multiplex, small fiber neuropathy
Clinical Features
Cranial neuropathies (most common): - Facial nerve palsy (CN VII) - hallmark; often bilateral - Optic neuropathy (CN II) - vision loss, papilledema - Other cranial nerves: III, IV, VI (diplopia), VIII (hearing loss, vertigo)
Meningeal involvement: - Chronic meningitis - headache, cognitive impairment - Hydrocephalus from CSF obstruction
Parenchymal disease: - Focal neurological deficits - Seizures - Myelopathy (spinal cord involvement) - May mimic MS or CNS vasculitis
Hypothalamic-pituitary dysfunction: - Diabetes insipidus (most common endocrine manifestation) - Amenorrhea, galactorrhea - Hypothyroidism, hyperprolactinemia
Peripheral nervous system: - Mononeuritis multiplex - Small fiber neuropathy - Symmetric polyneuropathy (rare)
Diagnosis
Clinical criteria: - Requires combination of clinical, radiologic, laboratory, and histopathological findings - Multidisciplinary evaluation often necessary
Imaging: - MRI with gadolinium contrast (imaging of choice) - Leptomeningeal enhancement (linear, nodular) - Parenchymal lesions (white matter, hypothalamic) - Cranial nerve enhancement - Hydrocephalus
CSF analysis: - Lymphocytic pleocytosis (typically 10-100 cells/μL) - Elevated protein (50-200 mg/dL) - Elevated ACE levels (not specific) - Normal or low glucose - Negative infectious studies
Tissue biopsy (gold standard): - Non-caseating granulomas on histopathology - Usually from extraneuronal sites (lymph nodes, lung, skin) - Brain/meningeal biopsy if extraneuronal biopsy unavailable
Supportive tests: - Chest CT: hilar/mediastinal lymphadenopathy, pulmonary infiltrates - Serum ACE: elevated (not specific) - PET scan: increased FDG uptake in affected areas
Differential diagnosis: - Multiple sclerosis - Neurosyphilis - CNS lymphoma - Primary CNS vasculitis - Tuberculosis meningitis - Lyme disease - Behçet’s disease
Management
First-line therapy: - High-dose corticosteroids - Prednisone 1 mg/kg/day (typically 60-80 mg/day) - IV methylprednisolone 1 g/day × 3-5 days for severe cases - Gradual taper over months based on clinical/radiologic response
Steroid-sparing immunosuppressants: - For chronic disease, steroid dependence, or refractory cases - Methotrexate 15-25 mg/week (most commonly used) - Azathioprine 2-3 mg/kg/day - Mycophenolate mofetil 1000-1500 mg twice daily - Hydroxychloroquine 200-400 mg/day (adjunctive)
Biologic agents: - For severe or refractory disease - Infliximab (TNF-α inhibitor) - most evidence - Adalimumab - alternative TNF-α inhibitor
Symptomatic management: - Seizures: antiepileptic drugs as indicated - Endocrine dysfunction: hormone replacement (DDAVP for DI, thyroid hormone) - Hydrocephalus: VP shunt or EVD if needed - Pain: neuropathic pain management
Monitoring: - Serial MRI every 3-6 months initially, then annually - CSF re-evaluation if clinical worsening - Monitor for systemic disease progression - Screen for steroid complications
Prognosis: - Variable course: relapsing-remitting or chronic progressive - Early aggressive treatment improves outcomes - Regular follow-up essential due to recurrence risk
Multiple Choice Question
Which of the following is the most common neurological manifestation of neurosarcoidosis?
- Optic neuritis
- Facial nerve palsy
- Myelopathy
- Hypothalamic dysfunction
Answer: (B) Facial nerve palsy Cranial neuropathies, particularly facial nerve palsy is common in neurosarcoidosis.
References
- Fritz D, Voortman M, van de Beek D, Drent M, Brouwer MC. Many faces of neurosarcoidosis: from chronic meningitis to myelopathy. Curr Opin Pulm Med. 2017;23(5):439–446. DOI: 10.1097/MCP.0000000000000411
- Bradshaw MJ, Pawate S, Koth LL, Cho TA, Gelfand JM. Neurosarcoidosis: Pathophysiology, Diagnosis, and Treatment. Neurol Neuroimmunol Neuroinflamm. 2021;8(6):e1084. DOI: 10.1212/NXI.0000000000001084
- Stern BJ, Royal W 3rd, Gelfand JM, Clifford DB, Tavee J, Pawate S, et al. Definition and consensus diagnostic criteria for neurosarcoidosis: from the Neurosarcoidosis Consortium Consensus Group. JAMA Neurol. 2018;75(12):1546–1553. DOI: 10.1001/jamaneurol.2018.2295
Subarachnoid Hemorrhage (SAH)
- Incidence: 6-10 per 100,000/year; peak age 40-60 years
- Mortality: ~30% at 30 days despite treatment
- Main cause: Ruptured saccular (berry) aneurysm (85%)
- Classic symptom: “Thunderclap” headache - worst headache of life
- Key complication: Delayed cerebral ischemia from vasospasm
- Treatment: Nimodipine + early aneurysm securing (clip/coil)
- Thunderclap headache - sudden onset, peaks in seconds
- Sentinel headache - warning leak before major rupture
- Loss of consciousness at headache onset
- Nuchal rigidity with photophobia
- Terson’s syndrome - retinal hemorrhages (severe SAH)
- Seizures at presentation (increased ICP)
- Declining mental status - suspect rebleeding or hydrocephalus
Clinical Scenario
A 52-year-old woman presents to the emergency department with sudden, severe headache described as "the worst headache of my life." The pain began abruptly while lifting a heavy box, followed by brief loss of consciousness and nausea. On examination, she is drowsy, photophobic, and has mild neck stiffness but no focal neurological deficits. Non-contrast CT brain shows hyperdensity within the basal cisterns consistent with subarachnoid blood. This presentation is typical of aneurysmal rupture leading to acute subarachnoid hemorrhage.
Epidemiology
- Accounts for ~5% of all strokes but disproportionately high mortality
- Incidence: 6-10 cases per 100,000/year; peak age 40-60 years
- More common in women; risk factors: smoking, hypertension, family history
- 85% due to ruptured saccular (berry) aneurysm (anterior circulation most common)
- One-third die within 30 days; many survivors have long-term deficits
Aneurysmal rupture: - Most non-traumatic SAH from ruptured intracranial saccular aneurysm - Blood enters subarachnoid space → sudden ICP rise → transient global cerebral ischemia - Blood breakdown products trigger vasospasm, inflammation, delayed cerebral ischemia (DCI) - Hydrocephalus from CSF reabsorption obstruction at arachnoid granulations
Less common causes: - Arteriovenous malformations (AVMs) - Mycotic aneurysms - Vasculitis - Idiopathic perimesencephalic hemorrhage (better prognosis)
Complications: - Rebleeding (highest risk first 24 hours) - Vasospasm (peaks days 3-14) - Hydrocephalus (acute or delayed) - Seizures, cardiac arrhythmias
Clinical Features
Classic presentation: - Thunderclap headache - sudden, severe, peaks within seconds - Nausea, vomiting, photophobia - Transient or prolonged loss of consciousness
Examination findings: - Nuchal rigidity (develops hours after onset) - Positive Kernig’s or Brudzinski’s signs (meningeal irritation) - Focal deficits if intracerebral extension or vasospasm-related ischemia - Seizures may occur - Terson’s syndrome (retinal hemorrhages) in severe cases
Diagnosis
Initial workup: - Non-contrast CT head - first test; detects SAH in >95% within 24 hours - If CT negative but high suspicion: lumbar puncture showing xanthochromia (gold standard) - CT angiography (CTA) or MR angiography (MRA) - identify bleeding source - Digital subtraction angiography (DSA) - gold standard for aneurysm detection
Grading scales: - Hunt-Hess or WFNS scales - guide prognosis and management
Differential diagnosis: - Migraine with aura - Meningitis - Reversible cerebral vasoconstriction syndrome (RCVS) - Intracerebral hemorrhage - Venous sinus thrombosis
Management
Acute phase: - Airway protection, ICU monitoring - Blood pressure control (systolic BP <160 mmHg pre-securing; avoid hypotension) - Nimodipine 60 mg PO q4h × 21 days - prevent delayed cerebral ischemia/vasospasm - Prevent rebleeding: bed rest, analgesia, avoid Valsalva
Definitive treatment: - Aneurysm securing within 72 hours: surgical clipping or endovascular coiling - Choice depends on aneurysm location, patient factors, institutional expertise
Complications management: - Hydrocephalus: external ventricular drainage (EVD) - Vasospasm/DCI: maintain euvolemia (isotonic fluids), avoid hypovolemia - Rebleeding: early aneurysm treatment - Seizure prophylaxis: consider short-term anticonvulsants
General measures: - Glucose control, normothermia - ICP monitoring if indicated - DVT prophylaxis (after aneurysm secured)
Multiple Choice Question
Which of the following is the most sensitive investigation for detecting subarachnoid hemorrhage when CT scan is negative and suspicion remains high?
- Magnetic resonance imaging (MRI) of the brain
- Lumbar puncture demonstrating xanthochromia
- CT angiography
- Electroencephalography (EEG)
Answer: (B) Lumbar puncture demonstrating xanthochromia.









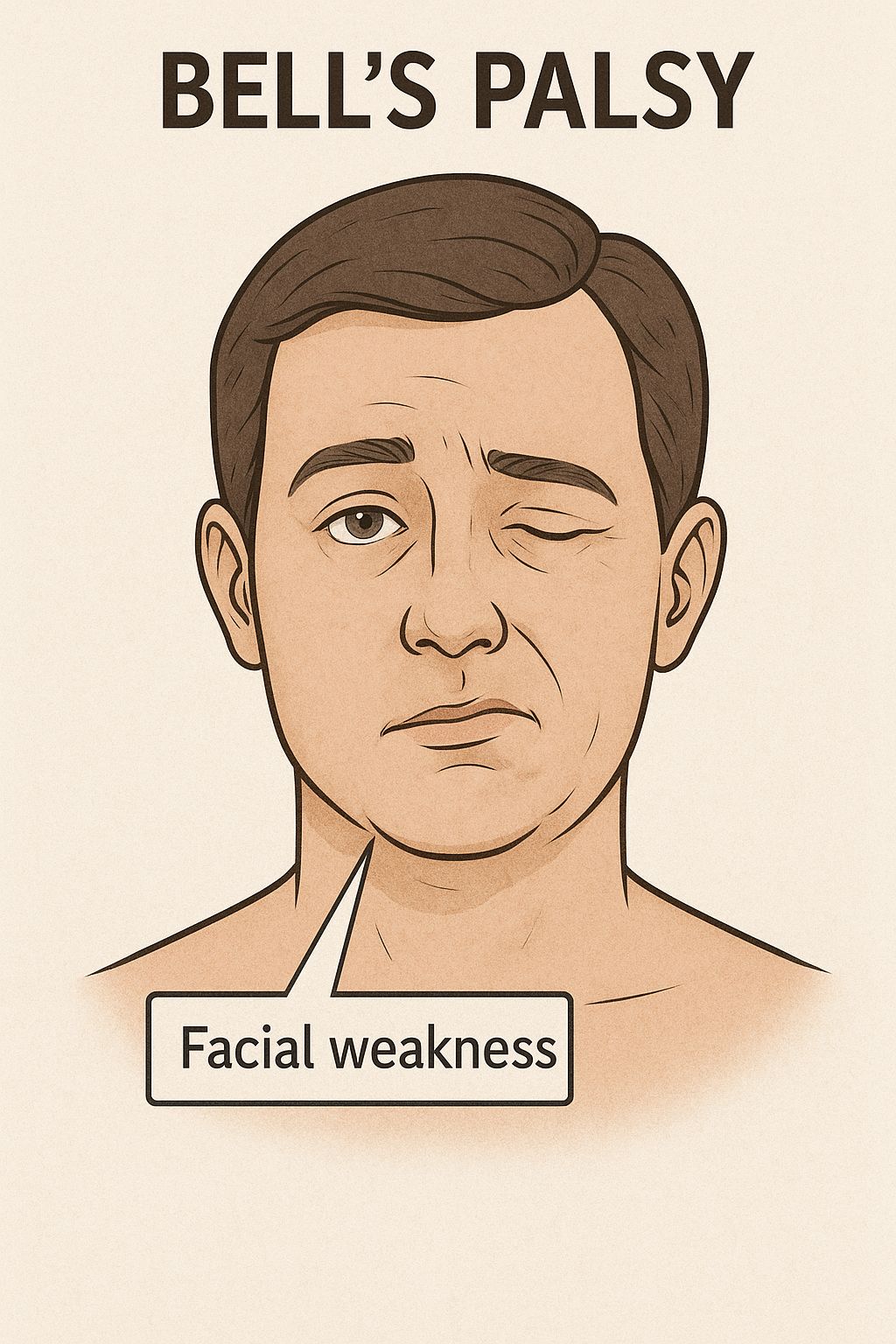

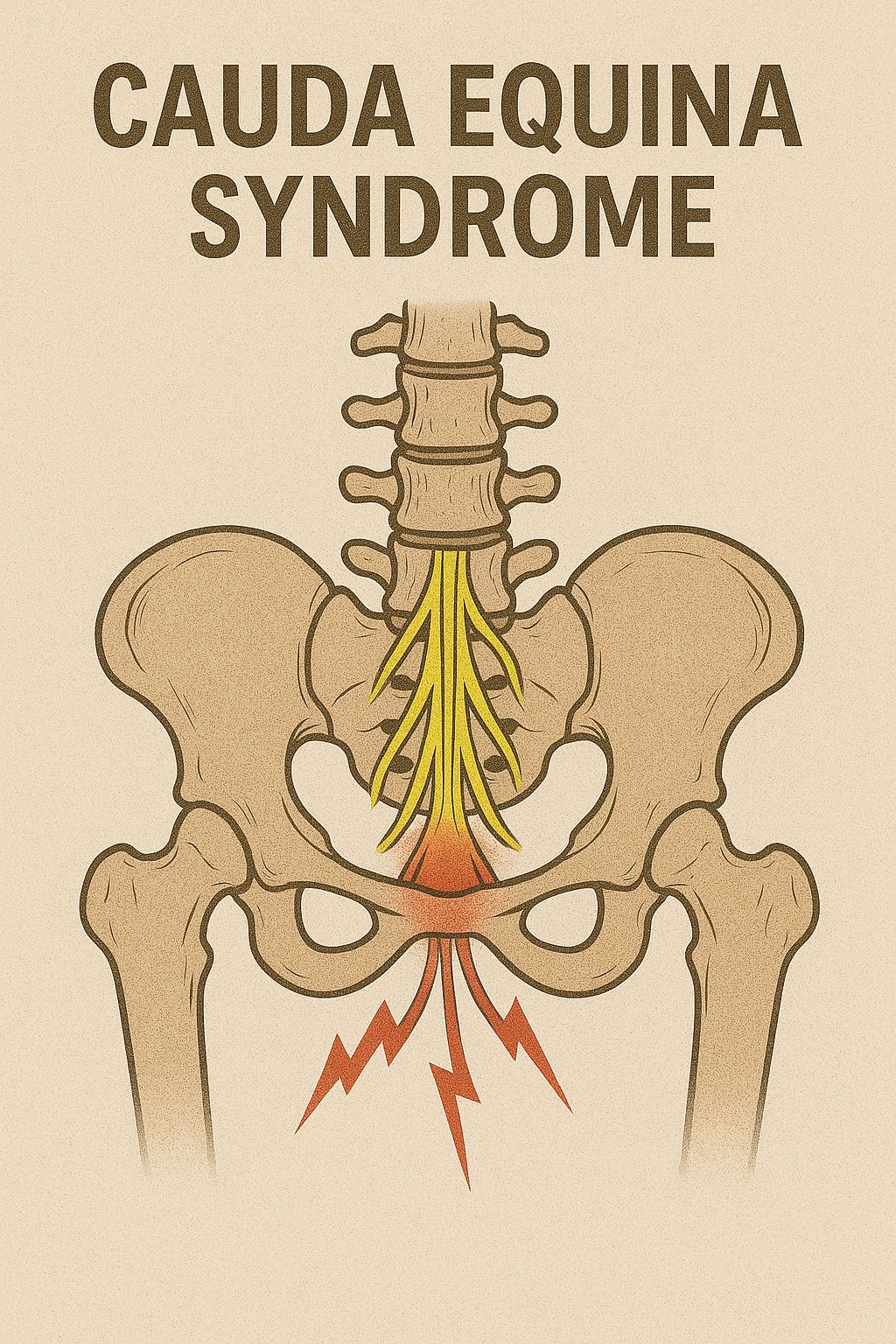



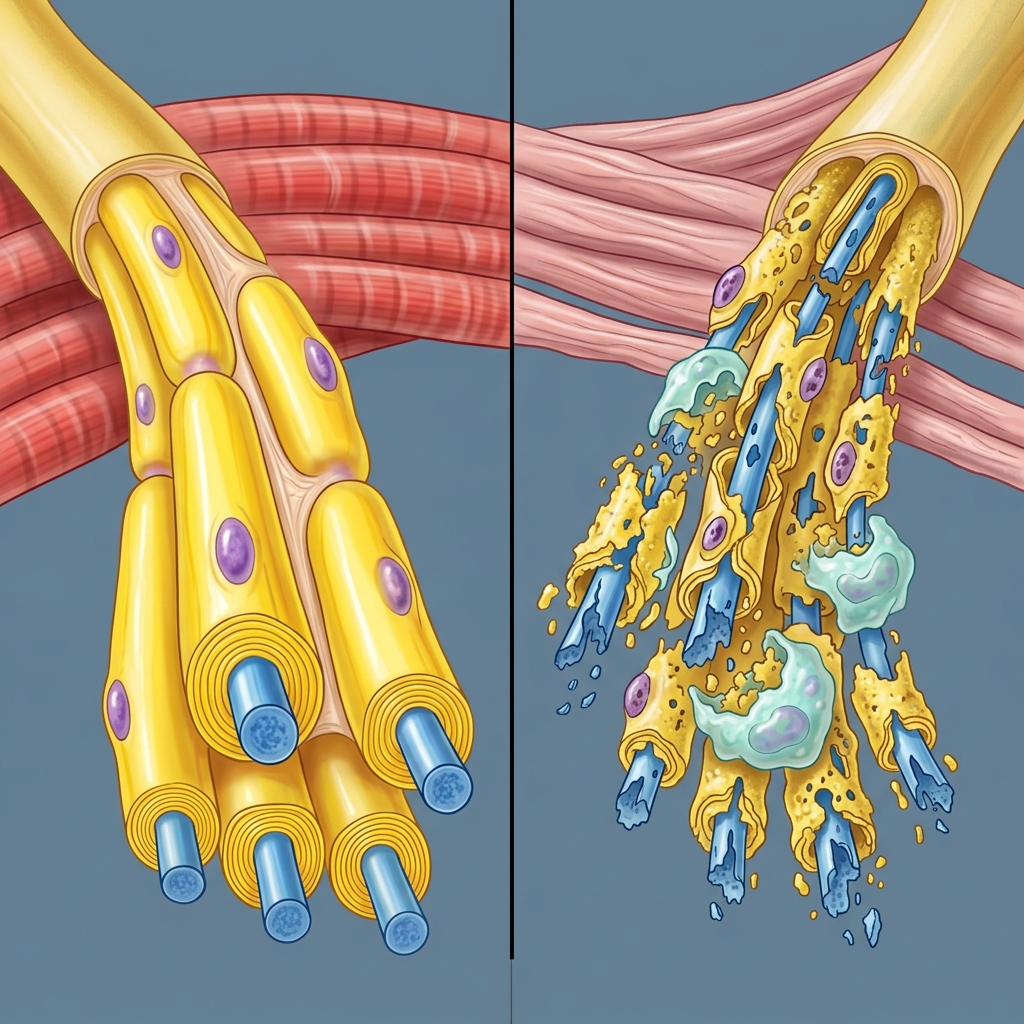
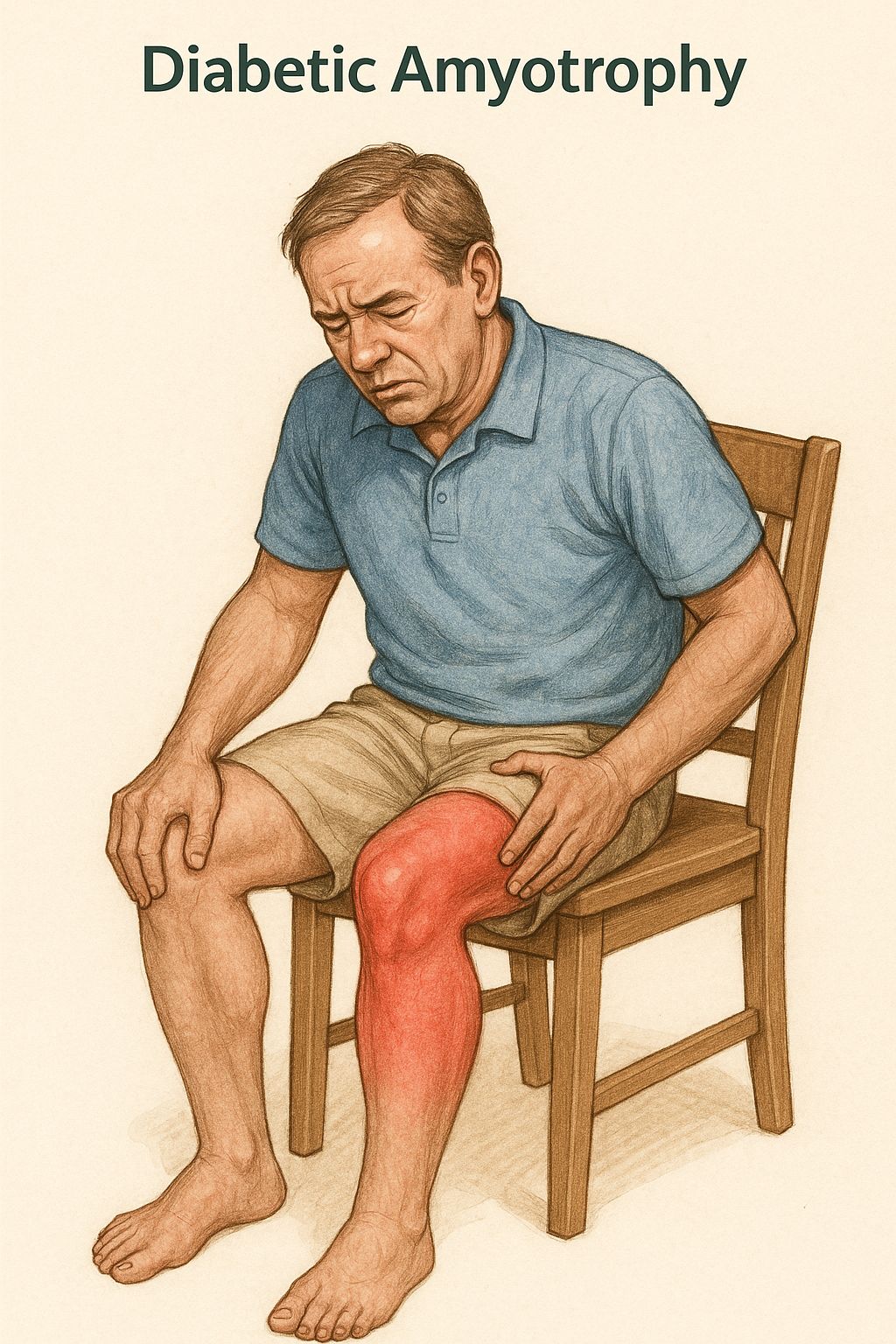

 Figure 1: Axial FLAIR MRI showing right temporal lobe hyperintensity consistent with herpes simplex encephalitis.
Figure 1: Axial FLAIR MRI showing right temporal lobe hyperintensity consistent with herpes simplex encephalitis.